Hipertensiunea arterială (HTA) este una
dintre cele mai frecvente boli cronice, afectând peste un miliard de persoane
în întreaga lume1.
Ca urmare a riscului crescut de complicaţii asociate (accidente vasculare, boală
coronariană şi boală renală cronică), HTA reprezintă o sursă majoră de
morbiditate şi mortalitate. Prevalenţa înaltă a HTA şi consecinţele sale au
deci un impact economic negativ semnificativ, individual şi populaţional, care
evidenţiază importanţa prevenţiei primare
a HTA (7).
În plus, cu toate progresele terapeutice
recente, controlul HTA este deseori
nesatisfăcător şi pentru mulţi bolnavi tratamentul aplicat nu realizează
nivelurile ţintă ale tensiunii arteriale (TA) stabilite de ghidurile actuale.
Pentru majoritatea bolnavilor cu HTA esenţială nu pot fi determinate cauzele creşterii TA, iar stabilirea
unui „profil individual“ al bolii
este dificilă, făcând practic imposibilă alegerea
unei terapii optime pentru un anumit bolnav şi stabilirea unui prognostic;
în consecinţă, tratamentul antihipertensiv este eminamente empiric, bazat pe
patogenia bolii, contextul epidemiologic şi trialurile clinice (1). În aceste
condiţii, înţelegerea mai bună şi, pe cât posibil, mai completă a patogeniei
moleculare a HTA rămâne o prioritate.
Determinanţii principali ai TA sunt fluxul sanguin (dependent de debitul
cardiac şi volumul sanguin) şi rezistenţa
vasculară periferică (produsă de starea de contracţie a arterelor mici şi
arteriolelor)2.
Ambele componente sunt reglate printr-o reţea complexă, intricată, de căi
fiziologice care asigură – prin mecanisme renale, neurovegetative şi endocrine
– homeostazia volumului fluidului extracelular, contractilitatea cardiacă şi
tonusul vascular (1, 7). Perturbarea oricăreia din aceste căi fiziologice –
produsă de factori de mediu şi de factori genetici – va avea ca efect creşterea
sau scăderea presiunii sanguine.
Fără a intra în descrierea mecanismelor de
reglare ale TA, accesibile în orice manual sau în articole de sinteză (6), vom
preciza doar câteva descoperiri recente în căile patogenice „clasice“, precum şi
identificarea unor căi noi (1):
• confirmarea rolului crucial al rinichilor în reglarea TA şi patogenia HTA, prin
intervenţia receptorilor AT1A pentru angiotensinogen
din tubii contorţi proximali în reglarea fenomenului de „natriureză presională“,
precum şi prin stimularea activităţii nervilor simpatici intrarenali asociată
cu creşterea reabsorbţiei de sodiu şi dezvoltarea HTA;
• descifrarea mecanismelor moleculare
implicate în rezistenţa vasculară
periferică, îndeosebi a căilor de semnalizare activate de receptorii
mediatorilor hormonali cuplaţi cu proteine G implicate în reglarea tonusului
vascular şi a TA;
• descoperirea rolului ţesutului interstiţial din piele ca „rezervor dinamic“ de
sodiu, care tamponează impactul acumulării de sodiu asupra volumului
intravascular şi TA;
• identificarea rolului important al inflamaţiei şi sistemului imun în dezvoltarea
HTA, care devine şi o boală autoimună (imunogenii endoteliali determină
infiltrarea şi activarea limfocitelor T în adventicea vasculară, urmată de
eliberarea unor citokine ce cresc TA).
Toate aceste descoperiri recente au
consecinţe terapeutice importante (1): blocarea receptorilor AT1, denervarea
renală (cu ajutorul unui cateter de radiofrecvenţă) în formele refractare de
HTA, blocarea căilor de semnalizare mediate de proteina G, dezvoltarea de
vaccinuri pentru HTA (faţă de imunogeni eliberaţi de endoteliul vascular sau
contra unor mediatori neurohormonali, de exemplu angiotensina II).
Deşi interacţiunile complexe dintre
sistemele care reglează TA au fost intens studiate în ultimele decenii, rolul
lor specific în producerea HTA nu este complet elucidat încă. Cert este doar
faptul că în aceste procese intervin numeroase molecule care sunt codificate de
gene şi a căror expresie (crescută sau redusă) este controlată prin mecanisme
genetice sau epigenetice. În acest context, vom sublinia din nou inabilitatea
cercetătorilor de a stabili cauzele HTA
la fiecare bolnav, inclusiv vulnerabilităţile sale genetice, fapt ce
constituie un obstacol major pentru dezvoltarea unui management clinic mai
personalizat şi mai eficient.
Existenţa unei componente genetice în
producerea HTA a fost sugerată de incidenţa familială crescută a bolii (chiar
în condiţiile în care diferiţi membri ai aceleiaşi familii trăiesc în medii
diferite, cu factori de risc variaţi), frecvenţa crescută la bărbaţi şi la
anumite grupuri etnice (de exemplu, afroamericani). Ereditatea HTA esenţiale a
fost mulţi ani un subiect de confruntare între două ipoteze (2): • Platt (1947)
a măsurat TA la persoane normotensive şi hipertensive precum şi la rudele lor,
găsind o distribuţie bimodală a TA
(bolnavii fiind o subpopulaţie distinctă faţă de normotensivi), fapt ce l-a
determinat să afirme că HTA este o boală monogenică mendeliană, produsă
de o singură mutaţie • Pickerring (1959) a demonstrat că TA are un caracter
cantitativ, complex, cu o distribuţie continuă (gaussiană) şi cu determinism poligenic; hipertensiunea (definită prin valori ale TA sistolice ≥
140 mm Hg)3 este numai porţiunea superioară (+ 2,5 deviaţii
standard) a curbei de distribuţie continue a TA. În timp, ipoteza Pickering a
fost confirmată de studii epidemiologice ample şi în prezent se consideră că marea majoritate a cazurilor de HTA sunt
determinate multifactorial, fiind produse de intervenţia mai multor gene de susceptibilitate ale
căror efecte sunt modulate de interacţiuni între diferite gene (epistazie) şi
între gene şi mediu. Totuşi, ipoteza Platt nu poate fi eliminată integral,
deoarece există forme rare de hipertensiune şi hipotensiune arterială care sunt
determinate de mutaţii monogenice rare, cu penetranţă înaltă şi efecte
semnificative. HTA prezintă cert o heterogenitate
fenotipică şi genotipică.
După stabilirea unui model etiologic
multifactorial, s-a încercat determinarea contribuţiei
relative a genelor în determinismul bolii. Mai multe studii recente au
estimat că ponderea eredităţii (numită heritabilitate)
în etiologia HTA este cuprinsă între 31 şi 68% (7), iar rudele de gradul I ale
bolnavilor cu HTA au un risc de trei-patru ori mai mare de a dezvolta boala
comparativ cu populaţia generală. Cu toate acestea, factorii de mediu (dieta, stilul
de viaţă, stresul, fumatul, alcoolul ş.a.) joacă un rol cauzal important,
conferind HTA titlul de „boală a civilizaţiei“.
Problema esenţială a geneticii HTA este identificarea genelor şi efectelor
diferitelor variante alelice ale acestor gene în modularea TA, precum şi
elucidarea mecanismelor genetice ce intervin în HTA. Rezolvarea acestor
elemente ar permite o înţelegere corectă şi completă a mecanismelor bolii,
stabilirea unui „profil patogenic
individual“ şi identificarea unor noi ţinte terapeutice. În pofida unor
eforturi ample, perfect justificate, această problemă este greu de rezolvat.
Studiul sindroamelor monogenice rare ce
afectează TA a permis identificarea unor mutaţii cu efecte majore în mai mult
de 20 de gene (7) ce produc modificări în: • excreţia renală de sodiu şi/sau
potasiu (de exemplu, sindroamele Bartter şi Gitelman, sindromul Gordon sau
sindromul Liddle) • sinteza steroizilor/aldosteronului (de exemplu, deficienţa
în 17a-hidroxilază sau hiperaldosteronismul familial) • sistemul simpatic (de
exemplu, paraganglioamele). Analiza acestor mutaţii
rare dar cu efecte mari a contribuit la înţelegerea mecanismelor de control
al TA. Cu toate acestea, marea majoritate a contribuţiei genetice în HTA
multifactorială a rămas neexplicabilă, datorită complexităţii bolii şi naturii
poligenice, care implică mai multe gene
care au fiecare efecte mici.
Identificare genelor de susceptibilitate implicate în HTA multifactorială se
bazează pe studiile de înlănţuire şi de asociere genică, care au ca scop
stabilirea unei asocieri semnificative a unei anumite regiuni cromozomiale sau
a unei variante alelice cu boala, pe baza ipotezei „boală comună – variantă comună“; această ipoteză porneşte de la
premisa că principalele alele de susceptibilitate vor putea fi identificate la
toţi bolnavii cu aceeaşi afecţiune (2, 3).
Analizele de înlănţuire s-au efectuat în familii de bolnavi pe baza prezumţiei că
bolnavii (de obicei fraţi afectaţi) vor avea mai frecvent în comun o anumită
alelă (a unei gene candidat) comparativ
cu persoanele sănătoase din aceeaşi familie. Studiile de asociere se efectuează într-o populaţie şi compară incidenţa unui anumit polimorfism genic la
bolnavi cu incidenţa lui într-un grup control; dacă o alelă specifică a
locusului polimorfic are o prezenţă semnificativ mai mare la indivizii cu boală
comparativ cu cei neafectaţi, atunci se poate presupune că alela respectivă
este implicată în patogenia bolii.
În ciuda complexităţii bolii şi dificultăţilor
tehnice, în perioada „pregenomică“ au fost identificate câteva variante genice
ce conferă susceptibilitate la HTA (2), în special ale genelor ce codifică
diferite componente ale sistemului renină-angiotensină,
canale ionice sau enzime ce participă la sinteza aldosteronului: • gena ADD1 (a-aducină, senzor al modificărilor
presiunii hidrostatice, situată pe cromozomul 4p16) • gena AGT (pentru angiotensinogen, situată pe cromozomul 1q42-q43) • genaREN (pentru renină, situată pe
cromozomul 1q32) • gena ACE (pentru
enzima de conversie a angiotensinei, situată pe cromozomul 17q23) • gena AT1R (pentru receptorul angiotensinei,
situată pe cromozomul 3q21) • gena GJA5
(pentru conexina 40, din structurile joncţionale ale celulelor endoteliale din
arteriola eferentă şi aparatul juxtaglomerular) • gena SCNN1B (pentru subunitatea b a canalului de sodiu non-voltaj dependent
tip 1, situată pe cromozomul 16p13-p12) • gena CYP11B2 (pentru aldosteron-sintetază, situată pe cromozomul 8q21).
În ultimii ani, studiile de asociere
„caz-control“ (pe baza ipotezei „boală
comună – variantă comună“) s-au realizat la nivelul întregului genom (în
engleză „genome wide association studies“
– GWAS)4, accelerând
descifrarea arhitecturii genetice a TA şi HTA. Folosind genotiparea prin
tehnici cu „debit mare“ (microreţele/microcipuri ADN de tip SNPs), se pot
cerceta printr-o singură scanare a genomului uman circa un milion de markeri
genetici care acoperă larg întregul genom. Genotiparea a zeci de mii de
pacienţi cu aceeaşi boală şi a unui lot control de indivizi sănătoşi a permis identificarea regiunilor genomice, asociate semnificativ cu boala, în care
sunt localizate variantele genelor de susceptibilitate la boală. Descoperirea
variantei genice cauzale (în regiunea cromozomială identificată) şi determinarea
mecanismului prin care această variantă participă la realizarea bolii necesită,
evident, alte studii: • cartografierea
de fineţe pentru reducerea la maximum posibil a dimensiunii regiunii în
care s-ar putea găsi gena de susceptibilitate • analiza secvenţelor ADN în
intervalul minim determinat, pentru identificarea genei/genelor prezente,
fiecare cu numeroase variante alelice • testarea
funcţională a variantelor genelor candidat pentru a determina, pe cale
experimentală, efectul funcţional al genei identificate şi rolului ei în
patogenia bolii (4).
În perioada 2008–2011, au fost realizate,
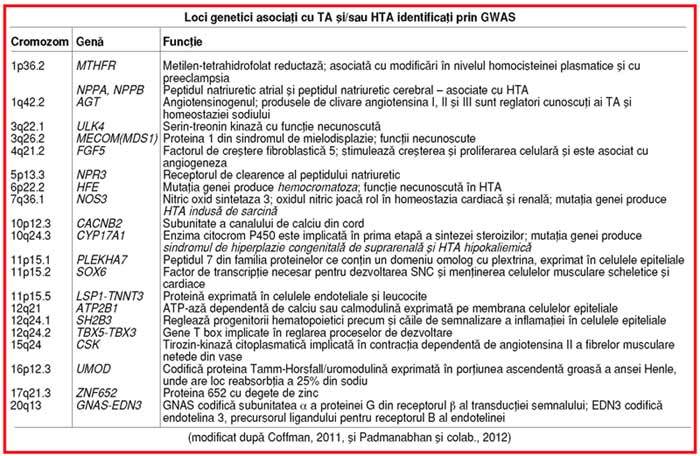
pe populaţii largi şi cu origini etnice diferite, şapte studii GWAS pentru TA şi/sau
HTA (1, 7); au fost identificate 41 de SNPs asociate semnificativ cu TA şi/sau
HTA. În regiunile cromozomiale (˜ 100
Kb)/locii definite prin aceste „semnale“, identificarea
unor gene cauzale s-a dovedit a fi dificilă (v. tabelul); numai câteva gene (NPPA, NOS3, UMOD) sunt asociate clar cu un anumit SNP, situat în
secvenţa unei gene; majoritatea SNPs se află în regiuni bogate în gene şi
atunci s-a luat în calcul „gena cea mai apropiată“ (v. tabelul); alteori, SNPs pot implica „secvenţe reglatoare“
(situsuri de fixare a unor factori de transcripţie sau molecule de microARN)
pentru gene de risc care se află mai la distanţă şi care urmează a fi
identificate.
Analiza locilor identificaţi prin GWAS
pentru TA şi/sau HTA relevă câteva elemente
surprinzătoare (1, 7):
• numai două
dintre genele asociate cu HTA multifactorială sunt implicate şi în sindroamele
monogenice cu modificări ale TA: CYP17A1
în deficienţa în 17a-hidroxilază şi NOS3
în HTA indusă de sarcină (v. tabelul);
• majoritatea
locilor nu au conexiuni evidente cu căi patogenice cunoscute a fi implicate
în HTA; excepţie fac: gena CYP17A1 –
ce participă la sinteza steroizilor, inclusiv a aldosteronului; genele NPPA, NPPB şi NRP3 (pentru peptidele
natriuretice şi receptorul lor) sau genaAGT, pentru angiotensinogen – implicate în echilibrul electrolitic
renal; gena EDN3 ce codifică o
endotelină – care are funcţii vasculare; restul locilor asociaţi cu HTA
participă la alte căi patogenice, fie necunoscute, fie nou identificate (v. mai jos);
• unii loci asociaţi cu TA şi/sau HTA sunt asociaţi puternic şi cu alte boli
comune, de exemplu, boala cronică renală, boala coronariană, anevrismele
intracraniene, schizofrenia ş.a., fapt ce impune cu necesitate efectuarea unor
studii funcţionale pentru a stabili rolul genelor asociate; una din explicaţiile
posibile ar fi implicarea lor în relee
patogenice comune unor boli diferite.
GWAS în TA/HTA au evidenţiat cel puţin două posibile căi patogenice noi. Prima
implică gena UMOD care prezintă o
variantă alelică asociată cu un risc scăzut de HTA: creşterea secreţiei de
uromodulină scade sensibilitatea maculei
densa la Cl– şi produce creşterea ratei de filtrare
glomerulară şi reducerea TA. A doua cale patogenică implică genele NOTCH şi JAG1, identificate recent în regiuni asociate cu HTA; calea de
semnalizare NOTCH participă la dezvoltarea sistemului cardiovascular şi, prin
mecanisme necunoscute, la reglarea TA (1).
Rezultatele obţinute prin GWAS demonstrează
evident că există numeroase variante
genetice ce influenţează TA/HTA în populaţii diferite. Totuşi, fiecare
dintre locii identificaţi prezintă numai un efect
foarte mic asupra variaţiei TA. Efectul combinat al tuturor locilor explică
doar 2% din heritabilitatea HTA (estimată între 31 şi 68%). În aceste condiţii,utilitatea predictivă a variantelor
asociate cu HTA (fie luate individual, fie incluse în scoruri de risc) este limitată.
Pentru a explica „heritabilitatea lipsă“ s-au formulat mai multe ipoteze (3): • nu
au fost „capturate“ variante structurale genomice „non-SNP“ (variaţii ale numărului
de copii – CNVs, sau inserţii şi deleţii – indels) care sunt frecvente şi ar
putea juca un rol în susceptibilitatea şi/sau patogenia bolii • GWAS nu poate
identifica variante rare (cu o frecvenţă populaţională sub 5%) dar cu efecte
mari şi nici interacţiuni între gene (epistazie) sau între gene şi mediu
(efecte epigenetice). Aceste critici sunt îndreptăţite, deoarece existenţa unorvariante genotipice rare dar cu efecte
mari a fost demonstrată printr-un studiu recent efectuat pe 5.000 de
persoane (incluse şi în Framingham Heart
Study), la care s-au analizat trei gene – SLC12A3, SLC12A1 şi KGNJ1 – asociate cu sindroamele monogenice
Bartter şi Gitelman, caracterizate prin reducerea TA (1); în cohorta Framingham
au fost identificate variante alelice rare ale acestor gene, asociate cu scăderea
TA şi un risc scăzut de HTA. De asemenea, modularea
epigenetică a expresiei unor gene implicate în reglarea TA şi HTA – ce
implică intervenţia unor factori de mediu şi unor mecanisme moleculare
specifice (modificarea histonelor, metilarea ADN sau acţiunea unor molecule de
micro ARN) – a fost demonstrată de două studii recente (7).
• Primul studiu a evidenţiat că infecţia cu
virusul citomegalic uman (HCMV) se asociază pozitiv cu HTA esenţială,
independent de alţi factori de risc; HCMV codifică un microARN care este
puternic exprimat la pacienţii hipertensivi; acest fapt a fost confirmat
experimental: infecţia şoarecilor cu CMV murin creşte TA.
• Al doilea studiu se referă la mecanismul
epigenetic al HTA induse de sare (ingestia de sodiu produce, prin creşterea
activităţii simpatice, retenţia de sodiu şi creşterea TA). S-a demonstrat că
stimularea receptorilor b-adrenergici induce, prin modificarea histonelor,
supresia transcripţiei genei WNK4,
care conduce la activarea cotransportorului sodiu-clor, retenţie de sodiu şi
creşterea TA.
Pentru a
depăşi limitele studiilor GWAS actuale se preconizează o nouă strategie,
care se bazează pe: • folosirea unor
versiuni îmbunătăţite de analiză genomică, ce evaluează o gamă largă de
polimorfisme structurale • creşterea dimensiunii loturilor analizate (prin
implicarea unor consorţii internaţionale) – care să permită identificarea unor
variante rare • utilizarea secvenţierii regiunilor genomice asociate cu HTA
pentru identificarea precisă a genelor • cuplarea unor studii de genomică funcţională
(3, 4).
Ce concluzii
se pot desprinde din studiile recente de analiză genomică a HTA? Cu toate
eforturile şi progresele înregistrate, arhitectura genetică complexă a HTA s-a
dovedit a fi mai dificil de descifrat decât poate ar fi sugerat heritabilitatea
sa importantă. Numeroase gene de susceptibilitate identificate au funcţii noi
în căi patogenice cunoscute sau intervin probabil în căi patogenice noi.
Elucidarea acestor aspecte necesită alte studii, dar va duce cu certitudine la
ameliorarea diagnosticului etiologic, stabilirea unui profil individual al
bolii şi optimizarea terapiei prin găsirea unor „soluţii personalizate“. În
sprijinul acestei idei ne vom referi, în încheiere, la farmacogenomica
medicamentelor antihipertensive.
În pofida existenţei a numeroase clase de
medicamente eficace şi a multor tipuri de medicamente din fiecare clasă, ratele
de control al TA printre hipertensivi sunt dezamăgitoare (sub 35%), în
principal datorită eficienţei reduse la unele persoane; în plus, răspunsurile
terapeutice la un medicament sau la o combinaţie de medicamente diferă mult
între bolnavi, sunt foarte variabile (5). Soluţia acestui inconvenient este individualizarea terapiei pe baza
informaţiei genetice personale sau, pe scurt, farmacogenomica hipertensiunii. Până în prezent s-au identificat
polimorfisme în mai multe gene implicate în acţiunea unor medicamente, care
explică variaţiile individuale ale efectelor lor: gena ACE (codifică angiotensinogenul) şi inhibitorii de angiotensină;
gena ADD1 (codifică a-aducina, senzor
de presiune hidrostatică) şi diureticele tiazidice; gena ADRB1 (codifică receptorul adrenergic b1) şi beta-blocante sau
gena KCNMB1 (codifică subunitatea
beta a canalului de potasiu activat de calciu) şi medicamente anticalcice sau
betablocante. Totuşi, potenţialul clinic al farmacogenomicii HTA va trebui să
fie mai clar, mai precis, pentru ca practicienii să beneficieze de un test
rapid care să ofere informaţii despre utilizarea medicamentelor
antihipertensive din prima linie; se estimează că până în 2020 aceste
deziderate se vor realiza.
1OMS apreciază că în
2020 numărul bolnavilor hipertensivi va depăşi 1,5 miliarde în întreaga lume;
2Alţi determinanţi ai
TA sunt vârsta, greutatea corporală, dieta şi etnia;
3La acest nivel, „beneficiile unei acţiuni terapeutice le depăşesc
pe cele ale lipsei de acţiune“ (1)
4Detalii asupra
studiilor GWA au fost prezentate într-un articol anterior publicat în „Viaţa
medicală“ (2).