În genetica moleculară, prin genom se înţelege astăzi ansamblul informaţiei ereditare din
moleculele de ADN ale unei celule/organism, situată în marea ei majoritate
(99,5%) în cromozomii din nucleu, dar şi în mitocondrii (0,5%); există deci un genom nuclear, mare şi complex, şi un genom mitocondrial, mic şi simplu.
Uzual, termenul de genom uman se foloseşte cu precădere pentru ADN nuclear şi, în
toate articolele publicate până acum, ne-am referit la acest genom. Pentru o
imagine corectă a medicinii genomice, este însă necesar să prezentăm şi genomul mitocondrial, nu numai datorită
funcţiilor multiple ale mitocondriilor în celule, ci mai ales datorită implicaţiilor
frecvente şi complexe pe care disfuncţiile mitocondriale le produc în patologia
umană.
Toţi ştim că mitocondriile sunt „uzina energetică“ a celulei, deoarece, în
marea majoritate a tipurilor celulare, ele convertesc eficient energia chimică
din alimente în ATP, necesar funcţiilor celulare. Dar mitocondriile joacă, de
asemenea, roluri-cheie în alte procese
celulare importante: controlul ciclului celular şi al proliferării,
termogeneza adaptativă, homeostazia calciului, răspunsul imun înnăscut, iniţierea
apoptozei, sinteza pirimidinelor, porfirinelor şi hemului, diverse procese
metabolice, detoxificarea amoniacului şi a radicalilor liberi etc. Disfuncţiile mitocondriale au fost
observate iniţial în boli mitocondriale monogenice rare, ulterior şi în
diferite stări patologice frecvente, cum ar fi: unele boli neurodegenerative,
cancer, diabet, boli cardiace, epilepsie, obezitate ş.a., precum şi ca răspuns
la acţiunea unor toxine externe, a infecţiilor virale sau a unor medicamente1.
În plus, declinul progresiv al expresiei genelor mitocondriale este o
caracteristică centrală a procesului normal de îmbătrânire. Toate aceste
implicaţii au fundamentat un domeniu nou şi distinct de patologie umană
interdisciplinară, numit generic medicină
mitocondrială.
medicamente1.
În plus, declinul progresiv al expresiei genelor mitocondriale este o
caracteristică centrală a procesului normal de îmbătrânire. Toate aceste
implicaţii au fundamentat un domeniu nou şi distinct de patologie umană
interdisciplinară, numit generic medicină
mitocondrială.
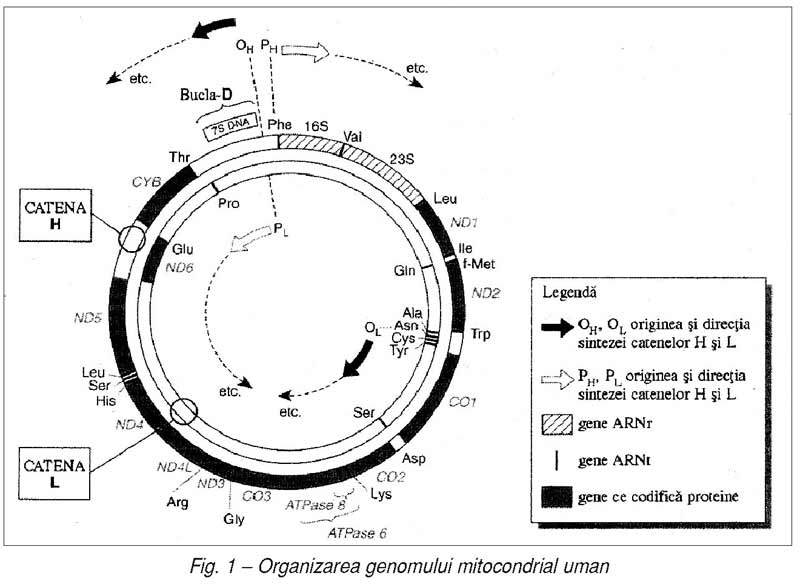
Celulele umane conţin între 500 şi 2.000 de
mitocondrii, fiecare dintre aceste organite având în alcătuirea sa două-zece
molecule circulare de ADN dublu catenar, formate din câte 16.569 pb. Cele două
catene de ADN mitocondrial (ADNmt)
se deosebesc prin conţinutul lor în baze
azotate (implicit, prin densitatea de plutire): una dintre catene este bogată înguanină (lanţul greu, H); în mod corespunzător, catena
complementară are un conţinut mare de citozină
(lanţul uşor, L) (fig. 1). Într-o mică regiune, ADNmt
este alcătuit din trei catene prin sinteza unei piese scurte numite ADN 7S sau bucla D (de la „displacement loops“), în care se află „centrii de reglare“ ai funcţiilor
ADNmt (originile replicării şi promotorii transcripţiei).
Secvenţa nucleotidică a ADNmt a fost complet
descifrată (Anderson şi colab., 1981), evidenţiindu-se câteva elemente
particulare genomului mitocondrial, diferite de genomul nuclear. Astfel, ADNmt nu este asociat cu proteine histonice
sau nehistonice şi aproape nu conţine ADNrepetitiv. Genomul mitocondrial este extrem
de compact: circa 93% din ADN
este format din secvenţe codante (absente doar în bucla D), ce formează 37 de gene: 13 gene codifică
polipeptide sintetizate în mitocondrii (constituenţi ai sistemului de
fosforilare oxidativă, prin care se produce majoritatea ATP celular), 22 de
gene codifică diferite molecule de ARNt (implicate în sinteza proteinelor
mitocondriale) şi două gene codifică ARNr (ce formează ribozomii
mitocondriali). Genele din ADNmt sunt „strâns împachetate“, aproape totdeauna contigue sau separate prin doar una-două
perechi de baze; ele nu conţin introni. Codul genetic
mitocondrial diferă puţin de cel
nuclear.
Multe dintre particularităţile genomului
mitocondrial prezintă similitudini cu
genomul procariotelor, fapt ce justifică ipoteza endosimbiotică, potrivit căreia ADNmt provine din ADN al
unor protobacterii aerobe primitive endocitate de strămoşii celulelor eucariote
de azi („arhea“), total dependente de
glicoliza anaerobă pentru a facilita producerea cantităţii necesare de molecule
macroergice ATP. Odată încorporate (eveniment petrecut în urmă cu circa 1,5
miliarde de ani), protobacteriile şi-ar fi pierdut autonomia, multe dintre
activităţile lor legate de generarea energiei fiind preluate şi controlate de către
genomul nuclear. Astfel, marea majoritate a proteinelor mitocondriale (multe
dintre ele fiind implicate în alte procese decât  sinteza de ATP) sunt
codificate de circa 1.000 de gene nucleare, fiind sintetizate în citoplasmă şi
importate în mitocondrii. Acest lucru este important de reţinut, deoarece
bolile mitocondriale pot fi determinate atât de mutaţii ale ADNmt, cât şi de
mutaţii ale genelor „mitocondriale“
din ADN nuclear.
sinteza de ATP) sunt
codificate de circa 1.000 de gene nucleare, fiind sintetizate în citoplasmă şi
importate în mitocondrii. Acest lucru este important de reţinut, deoarece
bolile mitocondriale pot fi determinate atât de mutaţii ale ADNmt, cât şi de
mutaţii ale genelor „mitocondriale“
din ADN nuclear.
Mutaţiile în ADNmt se produc cu o frecvenţă de 1020 de ori mai mare decât
în ADN nuclear, din cel puţin trei motive: (1) ADNmt nu formează complexe
cu histonele (cu rol protector) şi de aceea este deosebit de vulnerabil la acţiunea agenţilor mutageni,
dintre care unii – radicalii liberi de oxigen puternic agresivi – sunt generaţi
în cantităţi mari chiar în matricea mitocondrială, adică în mediul în care se
află moleculele de ADNmt; (2) În cursul replicării ADNmt, sub acţiunea
imperfectă a ADN-polimerazei gamma, se produc frecvent erori de împerechere a bazelor azotate; (3) Mitocondriile nu posedă sisteme eficiente de reparare a
ADN şi, ca atare, erorile inerente replicării, precum şi leziunile produse
accidental rămân, în marea lor majoritate, necorectate şi se acumulează în timp sub formă de mutaţii somatice, explicând
progresia bolilor mitocondriale, apariţia unor boli neurodegenerative la vârstnici,
precum şi procesul de senescenţă.
Trebuie subliniat faptul că genomul
mitocondrial al zigotului provine aproape exclusiv
din ovul, deci de la mamă2, fapt ce determină un tip particular de transmitere maternală a mutaţiilor
constituţionale (germinale) ale ADNmt şi a bolilor monogenice mitocondriale pe
care le produc: de la mamă la toţi copiii săi (de ambele sexe), în timp ce bărbaţii
bolnavi nu transmit boala (fig. 2).
Faptul că ADNmt este moştenit uniparental, exclusiv matriliniar, a permis
realizarea unor studii de filiaţie3, de „înrudire“ populaţională şi de
reconstituire a evoluţiei „pe linie maternă“ a populaţiilor umane, înapoi în
timp până la originea umanităţii (conceptul unui strămoş african comun pe linie
maternă, numit simbolic „Eva mitocondrială“),
sau a „călătoriilor continentale“ ale rudelor noastre ancestrale (fig. 3). Nu trebuie însă să uităm că
majoritatea proteinelor mitocondriale sunt sintetizate pe baza informaţiei unor
gene nucleare şi, în acest caz, transmiterea unor mutaţii4 se face clasic,
conform eredităţii mendeliene.
Atunci când se produce o mutaţie somatică,
dobândită, în ADN mitocondrial, rezultă în mitocondrie un amestec de molecule
mutante şi normale, numit heteroplasmie
(spre deosebire de prezenţa exclusivă a unui singur tip de mitocondrii, normal
sau mutant, numită homoplasmie). Când
o celulă se divide, ADNmt mutant va fi împărţit aleatoriu între celulele fiice şi astfel, în timp, procentajul de
ADNmt mutant între diferite linii celulare şi ţesuturi va fi diferit, fenomen
numit segregare replicativă. S-a
demonstrat că procentajul ADNmt mutant creşte în timp şi producţia de energie
scade treptat, până sub un prag minim
necesar funcţionării normale a ţesutului, moment în care manifestările clinice
devin evidente. De aceea, disfuncţiile mitocondriale prin mutaţii somatice au
un debut tardiv şi o evoluţie progresivă. Valoarea pragului energetic variază de la un ţesut la altul, cel mai
sensibil la deficitul energetic fiind sistemul nervos; urmează, în ordine, muşchii
scheletici, inima, rinichii, glandele endocrine şi ficatul.
Mai subliniem faptul că mitocondriile
prezintă o dinamică morfofuncţională
particulară, cu rol crucial în reglarea funcţiilor mitocondriale, reprezentată
de fisiunea (divizarea unei
mitocondrii în două sau mai multe entităţi independente) şi fuziunea mitocondrială (cuplarea şi
combinarea mai multor mitocondrii). Perturbările proceselor de fisiune şi
fuziune (determinate de mutaţii ale genelor nucleare care le guvernează) se
constituie în cauze ale unor patologii complexe, printre care figurează şi
unele afecţiuni neurodegenerative.
Prima boală
mitocondrială a fost identificată în urmă cu doar cinci decenii (Luft şi
colab., 1962) la un pacient cu „hipermetabolism“. Ulterior, au fost descrise
peste 200 de fenotipuri patologice atribuite alterării genetice a funcţiei
mitocondriale care, deşi au manifestări variate, prezintă o trăsătură unificatoare constituită
din deficitul balanţei energetice.
Scăderea, sub un prag critic, a producţiei de energie la nivel mitocondrial
antrenează perturbări majore ale activităţilor celulare, acumularea unor produşi
metabolici toxici (de exemplu, lactaţi), traduse prin declinul capacităţii unor
ţesuturi şi organe de a-şi îndeplini funcţiile specifice. Cele mai afectate
sunt sistemul nervos şi muşchii scheletici, a căror activitate este dependentă
prevalent de energia rezultată din procesul fosforilării oxidative
mitocondriale. Virtual însă, toate organele pot ajunge în stare de incapacitate
funcţională provocată de disfuncţii mitocondriale şi, ca atare, multe specialităţi medicale sunt inerent
confruntate cu acest tip de patologie. Pe lângă diversitatea şi
complexitatea lor, bolile mitocondriale sunt mult mai frecvente decât se estimează
de către unii clinicieni, incidenţa lor
depăşind cifra de 1:3.500 de nou-născuţi (valoare comparabilă cu aceea a
fibrozei chistice în populaţia europeană, considerată una dintre cele mai
frecvente boli genetice). Acest fapt implică, o dată în plus, necesitatea recunoaşterii şi diagnosticării
corecte a complexităţii patologiei mitocondriale, în contextul unui spectru
simptomatic foarte larg (manifestări multisistemice) şi a unei mari variabilităţi
clinice interindividuale (determinate de heteroplasmie, gene modulatoare etc.).
Vom preciza că, deşi unele explorări paraclinice (biochimice, histopatologice,
electromiografice etc.) pot documenta o posibilă disfuncţie mitocondrială, diagnosticul decisiv îl constituie analiza
moleculară a ADNmt.
Termenul de boli mitocondriale se referă de obicei la fenotipurile patologice
produse de mutaţii constituţionale ale ADNmt sau ale genelor nucleare ce
codifică proteine mitocondriale. Ele trebuie diferenţiate de disfuncţiile mitocondriale primare
determinate de acumularea unor mutaţii somatice, dobândite ale aceloraşi gene.
Aceste disfuncţii, care produc declinul producţiei de ATP, au fost descrise în
procesul de senescenţă, precum şi în diferite boli comune, cum ar fi: cancerul,
diabetul zaharat tip 2, cardiomiopatiile dilatative, epilepsia, obezitatea,
unele boli neurodegenerative (boala Parkinson, scleroza laterală amiotrofică,
boala Alzheimer) ş.a. Există şi disfuncţii
mitocondriale secundare produse de acţiunea unor factori externi (toxine,
infecţii virale sau numeroase medicamente) asupra sintezei sau funcţiei
proteinelor mitocondriale. În acest context, vom preciza că multe dintre reacţiile
adverse ce apar la medicamente frecvent folosite (statine, barbiturice, aspirină,
antiinflamatoare nesteroidiene, unele antibiotice, antidepresive,
antipsihotice, anticonvulsivante etc.) sunt consecinţa disfuncţiilor
mitocondriale. Se impune, aşadar, ca lansarea pe piaţă a unui medicament să fie
condiţionată de testarea prealabilă a toxicităţii sale asupra mitocondriilor,
pentru a proteja pacienţii de potenţialele efecte secundare, dar şi pentru a
elabora strategii nutriţionale capabile să reducă riscurile iatrogene.
Un domeniu de patologie „inaugurat“ acum şase
decenii a evoluat în ultimii ani într-o disciplină complexă, medicina mitocondrială. „Lecţiile“ învăţate
din studiile bolilor mitocondriale monogenice rare au implicaţii în
diagnosticul disfuncţiilor mitocondriale identificate în multe boli comune. În
câţiva ani, aplicarea noilor tehnologii de secvenţiere a exomului sau a întregului
genom vor produce o expansiune a cunoştinţelor despre mutaţiile genelor
nucleare în deficienţele mitocondriale primare, ce va permite dezvoltarea unor
mijloace terapeutice eficiente.