Frecvenţa,
diversitatea clinică şi complexitatea disfuncţiilor mitocondriale, constituţionale
sau dobândite, au fundamentat o disciplină nouă de patologie umană
interdisciplinară, numită generic medicina mitocondrială1. Au
fost descrise peste 200 de fenotipuri patologice atribuite alterării genetice a
funcţiei mitocondriale care, deşi au manifestări variate, prezintă o trăsătură
unificatoare constituită din deficitul balanţei energetice. Scăderea producţiei
de energie la nivel mitocondrial sub un prag critic antrenează perturbări
majore ale activităţilor celulare şi acumularea unor produşi metabolici toxici
(de exemplu, lactaţi), traduse prin declinul capacităţii unor ţesuturi şi
organe de a-şi îndeplini funcţiile specifice. Cele mai afectate sunt, firesc,
sistemul nervos şi muşchii scheletici dar, de fapt, toate organele pot ajunge în
stare de incapacitate funcţională. Disfuncţiile mitocondriale perturbă însă şi alte
roluri importante ale mitocondriilor în procesele celulare: controlul ciclului
celular şi al proliferării, iniţierea apoptozei, răspunsul imun înnăscut,
diferite sinteze şi procese metabolice etc.
Termenul
de boli mitocondriale se referă, de obicei, la fenotipurile patologice,
preponderent neuromusculare, produse de mutaţii constituţionale ale
ADNmt sau ale genelor nucleare ce codifică proteine mitocondriale. Ele trebuie
diferenţiate de disfuncţiile mitocondriale primare determinate de
acumularea, în timp, a unor mutaţii somatice, dobândite, ale aceloraşi
gene. Aceste disfuncţii, care produc declinul producţiei de ATP şi perturbarea
unor funcţii celulare, au fost descrise în procesul de senescenţă, precum şi în
diferite boli comune, cum ar fi: unele boli neurodegenerative, diabetul zaharat
tip 2, cardiomiopatiile dilatative, cancerul, epilepsia, obezitatea ş.a. Există
şi disfuncţii mitocondriale secundare produse de acţiunea unor factori
externi (toxine, infecţii virale sau numeroase medicamente) asupra sintezei
sau funcţiei proteinelor mitocondriale. Din această prezentare sintetică
rezultă că multiple specialităţi medicale sunt inerent confruntate cu
patologia mitocondrială, fapt ce implică necesitatea recunoaşterii şi
diagnosticării corecte a acestui tip de patologie, în contextul unui
spectru simptomatic foarte larg (manifestări multisistemice) şi a unei mari variabilităţi
individuale de manifestare şi de severitate.
Semnele
de debut ale bolilor mitocondriale pot apărea în orice moment al
ontogenezei, din perioada antenatală până la vârsta adultă, iar spectrul
simptomelor este foarte larg: manifestări oftalmologice, neuropatii periferice
sau afectări ale SNC, miopatii, disfuncţii hepatice, acidoză tubulară renală,
anomalii cardiace, endocrinologice şi/sau metabolice etc. După un interval de
timp relativ scurt, semnelor iniţiale li se adaugă – invariabil – cele ale afectării
neuromusculare. În general, semnele prezente la debut persistă şi se agravează
gradual, asociindu-se cu manifestări care exprimă suferinţa altor organe.
Principalele
elemente care sugerează implicarea alterării funcţiilor mitocondriilor în
etiopatogenia unei boli sunt: • debutul cel mai adesea precoce şi evoluţiarapidă şi progresivă • caracterul necoerent al asocierii semnelor
şi simptomelor • prezenţa în tabloul clinic a unor combinaţii de trăsături atipice,
inexplicabile • afectarea concomitentă a cel puţin trei organe care nu
au în comun nici originea embriologică şi nici funcţiile biologice • modificările
recurente survenite în tabloul clinic cu ocazia unor evenimente
precipitante (infecţii, efort, medicamente etc.). Dificultăţile diagnosticului
bolilor mitocondriale sunt determinate de efectele variate ale afectării de la
un organ la altul (în principal, din cauza fenomenului de heteroplasmie descris
în articolul anterior) şi de gradul diferit de severitate – de la un bolnav la altul.
Primul
pas important spre diagnosticul de boală mitocondrială îl reprezintă, ca de
obicei, un istoric atent şi complet (incluzând: manifestă ri ale tuturor
sistemelor, evenimente precipitante – mai ales folosirea anumitor medicamente,
istoric familial) urmat de un examen clinic detaliat (inclusiv sistemul nervos şi
organele de simţ, care uneori sunt „ignorate“). Tabloul clinic sugestiv pentru
o boală mitocondrială impune efectuarea unor examene paraclinice (nivelul seric
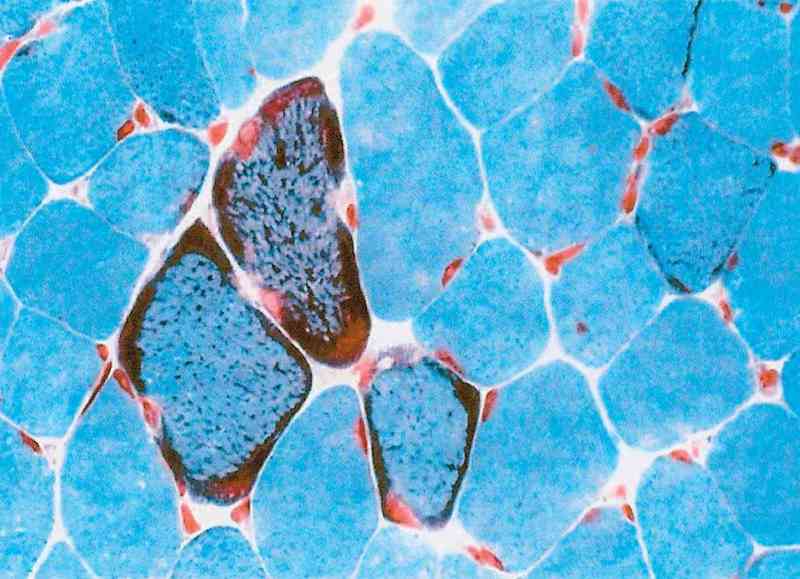
de piruvat, lactat, carnitină ş.a.; electromiografie; biopsie musculară: „fibre
musculare roşii“, depozite lipidice – v. figura) ale căror rezultate pot
documenta o posibilă disfuncţie mitocondrială. Testul decisiv de diagnostic îl
constituie însă analiza moleculară a ADNmt.
Există câtevacitopatii mitocondriale majore, bine definite molecular şi clinic:
oftalmoplegia externă cronică progresivă (CPEO – chronic progressive
external ophtalmoloplegia) şi forma sa severă, sindromul Kearns-Sayre
(KSS); epilepsia mioclonică cu fibre musculare roşii în lambouri (MERF – myoclonic
epilepsy with ragged-red fibres); sindromul MELAS (mitochondrial
myopathy, encephalopaty, lactic acidosis, stroke-like episodes); boala
Leigh (encefalomielopatie necrozantă subacută) şi sindromul NARP (neuropathy,
ataxia, retinitis pigmentosa); neuropatia optică ereditară Leber (LHON – Leber’s
hereditary optic neuropathy); sindromul miopatie cu oftalmoplegie externă,
neuropatie gastrointestinală, encefalopatie (MNGIE). Denumirea lor este
sugestivă pentru tabloul clinic, iar detalii pot fi obţinute în tratatele de
specialitate2 sau pe internet. Reamintim că bolile mitocondriale pot
fi produse de mutaţii constituţionale ale ADNmt (şi atunci se transmit exclusiv
pe linie maternă) şi/sau de mutaţii cu transmitere mendeliană ale genelor
nucleare, ce contribuie la funcţiile mitocondriilor (de exemplu, boala Leigh).
Bolile
mitocondriale produse de mutaţii constituţionale ale ADNmt sau ADN nuclear sunt
individual rare (în ansamblul lor, ele afectează însă peste 1:3.500 nou-născuţi).
Mult mai frecvente sunt însă disfuncţiile mitocondriale primare determinate
de mutaţii somatice, dobândite în cursul vieţii, ale genelor implicate în
funcţiile multiple ale mitocondriilor. În articolul precedent, arătam că mutaţiile
ADNmt se produc cu o frecvenţă de zece ori mai mare decât cele ale ADN nuclear,
datorită particularităţilor sale structurale, care îl fac foarte vulnerabil la
acţiunea radicalilor liberi de oxigen, generaţi în cantităţi mari chiar în
matricea mitocondrială. Aceste mutaţii variate (frecvent deleţii) se
acumulează în timp şi determină declinul progresiv al producţiei de ATP.
Dezechilibrul dintre necesarul de energie şi posibilitatea asigurării acestuia
a fost implicat în procesul de senescenţă şi se află la originea unor boli
degenerative ale vârstei a treia, precum şi a altor boli comune.
Senescenţase asociază cu diminuarea progresivă a masei musculare (sarcopenie) şi,
secundar, a forţei musculare, ceea ce compromite autonomia funcţională a
activităţilor motorii şi reduce calitatea vieţii. De reţinut că exerciţiile
fizice submaximale ameliorează slăbiciunea musculară asociată cu îmbătrânirea. În
schimb, acumularea mutaţiilor în ADNmt din neuronii SNC, în special din
ganglionii bazali, produce boli neurodegenerative greu de contracarat.
Boala
Parkinson (BP) este una dintre aceste afecţiuni cronice progresive, produsă
prin distrugerea treptată a neuronilor dopaminergici din sistemul
extrapiramidal (în special din ganglionii bazali), asociată cu depunerea unor
depozite proteice (corpi Lewi) şi depleţia de dopamină. Majoritatea cazurilor
sunt sporadice şi au un determinism multifactorial (ce implică existenţa unor
gene de susceptibilitate şi agresiunea unor factori externi); există însă 10–25%
de cazuri familiale determinate de mutaţii în genele SNCA, LRRK2, PARK2,
PARK7, PINK1 – ce codifică proteine cu rol în transmiterea sinaptică
(-sinucleina) sau funcţia mitocondriilor (dardarina, parkina, proteina DJ);
proteinele mutante suprimă capacitatea celulelor de a neutraliza radicalii
liberi, a căror acumulare se soldează cu moartea neuronilor dopaminerici.
Studiul formelor monogenice de BP a fundamentat rolul major al disfuncţiei mitocondriale
în această afecţiune şi introducerea terapiei cu levodopa.
Scleroza
laterală amiotrofică (SLA) este consecinţa degenerării motoneuronilor
somatici. Aproximativ 10% din bolnavi sunt purtători ai unor mutaţii autozomal
dominante cu penetranţă înaltă, exprimate la vârsta maturităţii. Circa o
cincime din cazurile familiale sunt consecinţa mutaţiilor care interesează gena
nucleară codificatoare a uneia dintre formele superoxid dismutazei (SOD1). În
lipsa SOD1, anionul superoxid – un radical liber generat în mitocondrii – rămâne
activ perioade mai lungi de timp şi îşi exercită efectul toxic asupra ADNmt şi
ADN nuclear, provocându-le leziuni ce se vor traduce prin deficit energetic.
În boala
Alzheimer (BA), mutaţiile ADNmt sunt detectabile în neuronii cortexului
cerebral, unde se înregistrează şi reduceri importante ale activităţii
piruvat-dehidrogenazei mitocondriale şi disrupţia proteinei mitocondriale Drp1 (dynamin-related
protein), provocată de oxidul nitric (un radical liber ce mediază
neurodegenerarea asociată cu BA). Sursa oxidului nitric o reprezintă peptidul beta-amiloid
din alcătuirea plăcilor senile. Aceste observaţii certifică participarea
perturbării mecanismelor mitocondriale generatoare de energie la patogenia
bolii. Identificarea proteinei Drp1 nitrozilate, ca element răspunzător de
alterarea sinapselor, se constituie în premisă a dezvoltării medicamentelor
care, blocând nitrozilarea Drp1, ar putea stopa sau încetini progresia BA.
Un alt ţesut
ale cărui funcţii sunt frecvent alterate ca urmare a acumulării defectelor
mitocondriale este miocardul. În fibrele musculare cardiace ale bolnavilor cu cardiomiopatie
dilatativă, analizele moleculare relevă prezenţa unor deleţii de dimensiuni
variate ale moleculelor ADNmt. Deoarece majoritatea cazurilor de cardiomiopatie
dilatativă sunt familiale, s-a sugerat că de producerea deleţiilor multiple ale
ADNmt se face răspunzătoare mutaţia unei gene nucleare cu rol în menţinerea
integrităţii genomului mitocondrial.
Diabetul
zaharat insulino-independent (DZ tip 2) debutează la adulţi şi vârstnici şi
manifestă trăsăturile unei boli degenerative. La unii bolnavi cu DZ tip 2, au
fost detectate mutaţii ale ADNmt în celulele insulelor Langerhans şi în fibrele
musculare scheletice. S-a demonstrat că agenţii diabetogeni (interleukina beta,
interferonul gamma, factorul de necroză tumorală tip alfa, aloxanul, streptozotocina)
– acţionează asupra mitocondriilor din celulele beta insulare, stimulând
producerea speciilor reactive de oxigen sau determinând alchilarea ADNmt.
Rupturile induse prin aceste mecanisme în moleculele ADNmt antrenează moartea
celulelor beta, urmată de instalarea insuficienţei producţiei de insulină.
O altă
problemă importantă este rolul mutaţiilor ADNmt în cancer. Există un
consens general asupra implicării genomului mitocondrial în iniţierea şi
promovarea cancerului. Mutaţiile ADNmt au fost identificate în majoritatea
tumorilor maligne şi fiecare tip de cancer analizat pare să conţină un model
caracteristic al alterărilor ADNmt, fapt ce sugerează că aceste mutaţii ar
putea fi o componentă esenţială în evoluţia tumorilor. Disfuncţia mitocondrială
în cancer scade producţia de ATP prin fosforilare oxidativă în mitocondrii; pentru
a realiza energia necesară proliferării celulare, se produce o reprogramare
a metabolismului energetic (caracteristică distinctivă a celulelor
tumorale), prin creşterea glicolizei aerobe în citosol. Disfuncţia mitocondrială
produce, de asemenea, creşterea producţiei radicalilor liberi de oxigen, care
sporesc rata mutaţiilor şi dereglează căile de semnalizare intracelulară,
importante pentru procesele de creştere şi diferenţiere celulară. O altă
consecinţă majoră a disfuncţiei mitocondriale în cancer este creşterea
rezistenţei în faţa mecanismelor de apoptoză şi activarea căilor de
supravieţuire celulară. O parte dintre aceste efecte ar putea fi „blocate“ prin
folosirea inhibitorilor glicolizei aerobe şi a unor compuşi proapoptotici.
Un ultim
aspect pe care îl semnalăm este reprezentat de disfuncţiile mitocondriale
secundare, produse prin acţiunea unor factori externi (toxine, infecţii
virale sau numeroase medicamente) asupra sintezei sau funcţiei proteinelor
mitocondriale. În acest context, vom preciza că multe dintre reacţiile
adverse ce apar la medicamente frecvent folosite (statine, aspirină,
antiinflamatoare nesteroidiene, barbiturice, unele antibiotice etc.) sunt
consecinţa disfuncţiilor mitocondriale. Se impune, aşadar, ca lansarea pe piaţă
a unui medicament să fie condiţionată de testarea prealabilă a toxicităţii sale
asupra mitocondriilor, pentru a proteja pacienţii de potenţialele efecte secundare,
dar şi pentru a elabora strategii nutriţionale capabile să reducă riscurile
iatrogene.
Diversitatea
clinică şi prevalenţa disfuncţiilor mitocondriale a fost doar recent recunoscută,
inaugurând domeniul „medicinii mitocondriale“. Diagnosticul acestor
disfuncţii necesită, în primul rând, „familiarizarea“ clinicienilor cu
patologia mitocondrială, pentru a creşte „gradul de suspiciune/alertă“ şi cu
explorările biochimice, histopatologice şi moleculare necesare pentru a
confirma observaţia de diagnostic.
Din păcate, nu există încă
tratamente eficace în disfuncţiile mitocondriale. Prevenirea unor factori
precipitanţi şi modificarea stilului de viaţă (exerciţii fizice, dietă săracă în
proteine şi bogată în lipide) pot fi benefice pentru bolnav, stimulând
biogeneza mitocondriilor. Opţiunile medicamentoase sunt limitate: administrarea
unor antioxidante (vitaminele A şi E, glutation, acid alfa-lipoic, coenzima Q10
şi forma sa sintetică, idebenon etc.), a unor compuşi ce îmbunătăţesc producţia
de ATP (vitaminele complexului B, în special B1 şi B2, levocarnitina, menadiona
etc.) sau care reduc formarea de lactat (dicloracetatul, piruvatul de sodiu). În
laboratoarele de cercetare se testează strategii noi bazate pe terapia genetică
(de exemplu, eliminarea moleculelor de ADNmt mutant prin blocarea replicării lor
cu oligonucleotide antisens) sau pe folosirea unor molecule mici, numite „cargo“,
ce pătrund specific, ţintit, în mitocondrii, transportând un efector (de
exemplu, un antioxidant – plastoquinonă). Aceste metode s-au dovedit eficiente,
dar introducerea lor în clinică implică depăşirea unor dificultăţi tehnice.
Cert este doar faptul că există speranţe autentice, dar… pentru mai târziu.