Patologia vasculară în otorinolaringologie
este deosebit de complexă, fiind reprezentată de afecţiuni specifice ale
capului şi gâtului sau de afecţiuni sistemice ce pot avea un răsunet important
în sfera ORL: epistaxisul, tumorile vasculare şi pseudovasculare, compresiunea
vasculară a nervilor cranieni, patologia vasculară în afecţiunile
cohleo-vestibulare, anomaliile vasculare ale gâtului, algiile vasculare ale
capului şi feţei, manifestările ORL ale vasculitelor.
Simptomatologia acestor afecţiuni este diversificată
în funcţie de localizarea şi de caracteristicile patologiei respective. Din
aceste considerente, investigaţiile paraclinice – imagistica (ecografie,
eco-Doppler) şi radiologia (angiografie, IRM, angio-IRM, CT) – joacă un rol
esenţial în diagnosticarea acestei patologii.
1.
Tumorile vasculare
Tumorile vasculare ale capului şi gâtului reprezintă
un grup heterogen de leziuni cu origine comună la nivelul sistemului vascular,
dar diferite din punct de vedere clinic, evolutiv şi terapeutic. Pot să apară
ca tumori izolate sau ca parte componentă a unor sindroame.
Există două tipuri de tumori: endoteliale
(benigne, maligne) sau perivasculare, dezvoltate prin proliferarea celulelor
de tip fibroblastic sau glomic (v.
tabelul)
HEMANGIOMUL
Hemangiomul
este cea mai frecventă tumoră vasculară benignă apărută la nivelul capului şi
gâtului, care poate afecta în egală măsură atât tegumentele, cât şi mucoasele.
În
aproximativ 30% din cazuri, hemangioamele sunt prezente încă de la naştere,
dar pot apărea şi la câteva săptămâni după naştere. Sunt tumori ce afectează în
special sexul feminin, raportul bărbaţi:femei fiind de 1:6.
Din
punct de vedere histopatologic, hemangioamele sunt tumori proliferative, ale căror
celule endoteliale prezintă o creştere a activităţii mitotice în timpul
proliferării.
Din
punct de vedere clinic, există trei tipuri de hemangioame: capilar, cavernos şi
mixt.
Hemangioamele
capilare se prezintă sub
forma unor zone tegumentare eritematoase prezente la naştere, fiind
caracterizate printr-o proliferare rapidă în primul an de viaţă, ulterior
începând să involueze. Hemangioamele ce afectează mucoasele pot fi localizate
la nivelul foselor nazale sau la nivelul laringelui (în special subglotic),
acesta din urmă afectând în special populaţia pediatrică. La nivelul foselor
nazale, hemangioamele se întâlnesc, cel mai frecvent, în treimea anterioară a
septului nazal, dar şi la nivelul cornetelor nazale şi a meaturilor. Clinic,
hemangioamele foselor nazale sau endosinusale se pot manifesta prin epistaxis
recidivant şi cefalee.
Hemangiomul
cavernos
se prezintă sub forma unor mase tumorale prost delimitate, compresibile, de
consistenţă moale, cu tendinţa de a implica structuri cu localizare profundă
(schelet, muşchi). Apariţia la nivelul maxilarului sau mandibulei este destul
de frecventă. În momentul în care hemangiomul laringian este diagnosticat la
adult, este vorba, cel mai frecvent, de forma cavernoasă cu localizare
supraglotică. Localizarea la nivelul mucoasei foselor nazale se poate manifesta
prin episoade repetate de rinoragie.
Hemangioamele
mixte se întâlnesc cu precădere la copii şi sunt localizate la nivelul
glandei parotide.
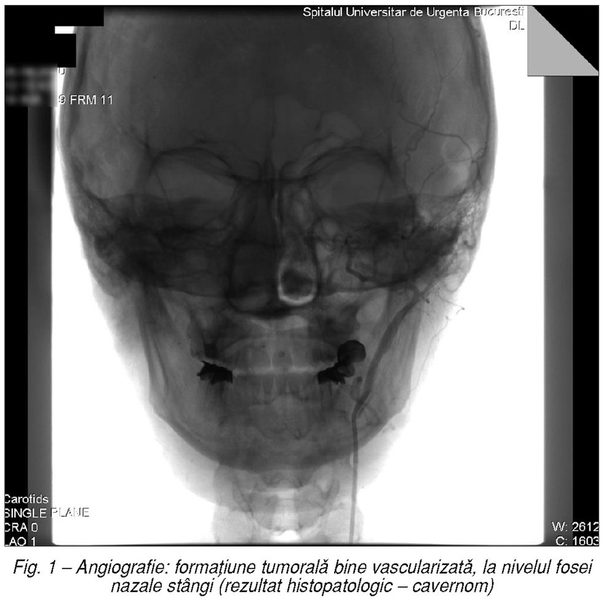
Protocolul de diagnostic cuprinde examenul
endoscopic, imagistica (CT, IRM) şi, nu în ultimul rând, angiografia cu sau fără
embolizarea pediculului vascular (fig.
1).
Examenul
endoscopic nazal poate evidenţia prezenţa unei formaţiuni tumorale roşiatice, uşor
sângerânde la atingere, situate în treimea anterioară a septului nazal, sau
leziuni roşii-violacee ce se întind pe mucoasa cornetelor nazale sau la
nivelul meatelor nazale, în special la nivelul meatului nazal mijlociu.
În
cazul localizării laringiene, examenul
laringofibroscopic evidenţiază prezenţa unei tumori submucoase situate în
special la nivelul peretelui posterolateral, subglotic.
Înaintea
alegerii unei strategii terapeutice, trebuie avut în vedere caracterul
involutiv al hemangioamelor capilare. Tratamentul de elecţie este cel
chirurgical. Dacă tumorile sunt unice şi de mici dimensiuni, se poate încerca
electrocoagularea. În cazul hemangioamelor cu localizare subglotică,
tratamentul poate consta în corticoterapie sistemică, injectare intralezională
de corticoizi sau excizie cu laser CO2.
LIMFANGIOMUL
 Limfangioamele
reprezintă malformaţii congenitale ale sistemului limfatic. Din punct de vedere
clinic şi histopatologic, limfangioamele pot fi: capilar, cavernos, higroma
chistică sau limfangiohemangiomul.
Limfangioamele
reprezintă malformaţii congenitale ale sistemului limfatic. Din punct de vedere
clinic şi histopatologic, limfangioamele pot fi: capilar, cavernos, higroma
chistică sau limfangiohemangiomul.
Limfangiomul
capilar
se prezintă sub forma unor papule la nivelul tegumentelor sau mucoaselor, cel
mai frecvent fiind localizat la nivelul limbii, precum şi subligual, la nivelul
podelei cavităţii bucale.
Limfangiomul
cavernos
poate fi confundat cu un lipom, manifestându-se sub forma unei mase tumorale
nedureroase, de consistenţă moale.
Higroma
chistică
are ca localizare predilectă regiunea posterioară a gâtului şi reprezintă o
malformaţie a vaselor limfatice, întâlnită în special în rândul populaţiei
pediatrice.
Limfangiohemangiomul reprezintă o tumoră
mixtă ce are caracteristicile limfangioamelor, dar care poate prezenta sângerări
episodice.
Clinic,
pacientul se poate prezenta pentru apariţia unor formaţiuni tumorale la nivelul
limbii sau la nivelul regiunii posterioare a gâtului, jenă dureroasă la deglutiţie,
disfagie şi chiar insuficienţă respiratorie.
Protocolul
de diagnostic cuprinde ecografia (utilă în determinarea relaţiei pe care formaţiunea
tumorală o are cu structurile vecine), imagistica (CT sau IRM – evidenţiază
impactul pe care formaţiunea tumorală îl are asupra structurilor învecinate),
iar puncţia cu ac fin poate ajuta la confirmarea diagnosticului.
Terapia
de elecţie este cea chirurgicală, cu excizie totală atunci când este posibil.
La copii, este de preferat efectuarea intervenţiei chirurgicale după vârsta
de 3–4 ani. Dacă excizia chirurgicală nu este completă, rata de recidivă este
de aproximativ 15%, majoritatea în primul an postoperator.
PARAGANGLIOAMELE
Paraganglioamele
sunt formaţiuni tumorale neuroendocrine cu originea la nivelul celulelor
parenchimatoase din neuroectoderm şi din crestele neurale. Sunt tumori
considerate benigne, dar pot avea un potenţial malign ridicat.
Celulele
care intră în alcătuirea paraganglioamelor pot elibera catecolamine,
colecistokinină, serotonină, somatostatină, VIP (peptidul intestinal
vasoactiv). Sunt celule asemănătoare celulelor din sistemul APUD (amine precursor uptake and
decarboxylation), sistem care înglobează şi reprezintă toate celulele
deviate din crestele neurale şi brahiomere sintetizatoare de polipeptide şi
amine fluorogene. Această caracteristică explică plurifocalitatea
paraganglioamelor, aspect ce poate explica existenţa mai multor paraganglioame
la acelaşi pacient.
Se
descriu următoarele tipuri: carotidian (glomus carotidian), timpanic (glomus
tympani), jugular (glomus jugulare), vagal (glomus vagal).
Paragangliomul carotidian
Glomusul
carotidian se dezvoltă de la nivelul chemoreceptorilor carotidieni situaţi în
peretele posteromedial al arterei carotide comune, la bifurcaţia acesteia.
acesteia.
Paragangliomul
carotidian poate avea caracter sporadic sau familial. Formele sporadice sunt
mult mai frecvente, iar în 10% din cazuri pot fi multicentrice, asociate cel
mai frecvent cu localizarea bilaterală. Formele ereditare sunt mai rare, 7–9%
din cazuri, dar cel mai frecvent sunt tumori multicentrice (30–40%). Glomusul
carotidian este paragangliomul cu cea mai mare capacitate de malignizare,
acest fenomen întâlnindu-se în 6–12% din cazuri. Afectează în mod egal ambele
sexe, fiind cel mai frecvent între 30 şi 60 de ani.
Clinic,
vorbim despre o tumoră cu evoluţie lentă, care poate să rămână mult timp
asimptomatică, fiind diagnosticată cel mai frecvent în a cincea decadă a vieţii.
Se prezintă sub forma unei formaţiuni tumorale nedureroase, situate anterior de
muşchiul sternocleidomastoidian, mobilă în plan lateral, dar fixată în plan
cefalo-caudal.
Simptomatic,
tumora poate genera apariţia unui suflu carotidian depistat la auscultaţie. Pe
măsură ce cresc în dimensiuni, tumorile glomice pot invada spaţiul
parafaringian, ducând la bombarea peretelui lateral al regiunii orofaringiene.
În aceste cazuri, pacientul poate prezenta disfagie, odinofagie sau sindroame
date de afectarea nervilor cranieni IX – XII.
Din
protocolul de diagnostic al glomusului carotidian face parte obligatoriu
imagistica. Examenele CT sau IRM efectuate cu substanţă de contrast sunt utile
în determinarea maselor tumorale (fig. 2).
Examenul
IRM permite evaluarea gradului de invazie vasculară a formaţiunii tumorale,
informaţie care ajută la alegerea tehnicii chirurgicale pentru fiecare caz în
parte şi la stabilirea prognosticului şi complicaţiilor:
Gradul I – tumoră situată la
nivelul bifurcaţiei arterei carotide comune, cu zonă de contact minim cu vasele
sanguine;
Gradul II – tumoră ce înglobează
aproximativ 50% din circumferinţa axului arterial principal. Formaţiunea
tumorală are un diametru mai mic de 5 cm;
Gradul III – tumoră ce înglobează
în totalitate axul arterial principal. Formaţiunea tumorală are un diametru mai
mare de 5 cm; III A – un segment al
arterei carotide interne este liber între polul superior al tumorii şi baza
craniului; III B – polul superior al
formaţiunii tumorale vine în contact cu baza craniului.
Angiografia (fig. 3) de arteră carotidă comună, cu sau fără embolizare,
indicată a fi efectuată bilateral, poate pune în evidenţă imaginea
patognomonică a glomusului carotidian: îngustarea bifurcaţiei arterei carotide
comune de către o formaţiune tumorală bine vascularizată (semnul „lirei“).
Terapia
de elecţie a glomusului carotidian este exereza chirurgicală, efectuată după embolizare prin
angiografie, şi reprezintă una dintre cele mai dificile din chirurgia
cervico-facială. În cazul glomusului carotidian bilateral, excizia celor două
formaţiuni tumorale trebuie făcută la o distanţă de trei până la şase luni.
Radioterapia,
singură sau asociată chirurgiei, este o altă strategie terapeutică ce poate fi
aleasă în cazul glomusului carotidian. Tratamentul se bazează pe capacitatea de
fibrozare locală, ce poate stopa evoluţia tumorală şi este indicată în cazurile
inoperabile sau în recidivele postoperatorii.
Paragangliomul jugulo-timpanic
Regiunea
jugulo-timpanică este punctul de plecare pentru două tipuri de paraganglioame:
timpanic şi jugular. Glomusul timpanic îşi are originea fie la nivelul nervului
vag (ramura auriculară posterioară a nervului Arnold), fie la nivelul nervului
Jacobson, ram al nervului glosofaringian. Glomusul jugular îşi are originea în
adventicea şi peretele venei jugulare.
Această
formaţiune reprezintă cele mai frecvente tumori de la nivelul urechii medii,
afectând în special sexul feminin, între 25 şi 65 de ani.
Clinic,
glomusul jugulo-timpanic prezintă o dezvoltare lentă, cele mai frecvente
simptome fiind hipoacuzia şi tinitusul pulsatil. Pacientul mai poate prezenta
senzaţie de ureche înfundată, otoree. Implicarea urechii interne poate duce la
apariţia crizelor vertiginoase şi a hipoacuziei neurosenzoriale pe audiograma
tonală.
 Apariţia
parezei nervilor cranieni IX – XI, sindromul de gaură ruptă posterioară
Vernet, caracterizat prin hemiparalizia şi hemianestezia vălului palatin, a
laringelui şi faringelui, este patognomonic.
Apariţia
parezei nervilor cranieni IX – XI, sindromul de gaură ruptă posterioară
Vernet, caracterizat prin hemiparalizia şi hemianestezia vălului palatin, a
laringelui şi faringelui, este patognomonic.Examinarea
otoscopică şi otomicroscopică evidenţiază prezenţa unei formaţiuni tumorale pulsatile,
roşie-albăstruie, situate înapoia membranei timpanice. Examinarea paraclinică
include evaluarea audiometrică (poate evidenţia o hipoacuzie de transmisie sau
neurosenzorială atunci când este afectată urechea internă), imagistica (CT –
eroziuni osoase, angio-IRM).
Tumori
cu mare potenţial eroziv şi extensiv, cazurile de glomus jugulo-timpanic pot fi
clasificate astfel (clasificarea lui Fisch):
Tipul A – tumoră limitată la nivelul
urechii medii (glomus timpanic);
Tipul B – tumoră limitată la regiunea
timpano-mastoidiană, fără afectarea compartimentului infralabirintic;
Tipul C – tumoră ce invadează
compartimentul infralabirintic al osului temporal, cu extensie spre apexul
petros: C1 – tumoră limitată la porţiunea
verticală a canalului carotidian; C2
– tumoră ce invadează porţiunea verticală a canalului carotidian; C3 – tumoră ce invadează porţiunea
orizontală a canalului carotidian;
Tipul D – extensie
intracraniană: D1 – mai mică de 2 cm
diametru; D2 – mai mare de 2 cm
diametru.
Diagnosticul
diferenţial trebuie făcut cu otita seroasă, colesteatomul, neurofibromul,
schwannomul, osteomul, otoscleroza, hemotimpanul idiopatic, proeminenţa
bulbului jugular.
Terapia
de elecţie, în cazul glomusului jugulo-timpanic, o reprezintă excizia chirurgicală.
Aceasta trebuie făcută în funcţie de gradul, localizarea şi extensia tumorii
(conform clasificării Fisch). Neuronavigaţia,
tot mai frecvent folosită, asigură, pe lângă o exereză extinsă a tumorii, o
protecţie foarte bună a structurilor vitale învecinate, un traumatism
chirurgical minim şi rezultate net superioare obţinute prin tehnicile clasice
pe cale deschisă.
Paragangliomul vagal
Paragangliomul
vagal este o formaţiune tumorală vasculară întâlnită cu o frecvenţă ridicată la
femei, în peste 70% din cazuri, care se dezvoltă de la nivelul celulelor
nervului vag. Aceste tumori se dezvoltă în apropierea originii muşchiului
sternocleidomastoidian şi se asociază cu pareza corzii vocale ipsilaterale. Pe
măsură ce creşte în dimensiuni, poate duce la apariţia sindromului Horner, prin
implicarea plexului faringian. Glomusul intravagal poate metastaza în 20% din
cazuri, cele mai frecvente fiind la nivel pulmonar.
Diagnosticul
diferenţial trebuie făcut cu tumorile neurale (neurofibroame, schwannoame) cu originea
la nivelul lanţului simpatic cervical.
Tratamentul
constă în excizia chirurgicală a formaţiunii tumorale, care se poate solda cu
sacrificarea nervului vag.
SINDROAMELE
VASCULARE
Sindromul
Osler–Weber–Rendu, cunoscut şi
sub denumirea de teleangiectazia ereditară hemoragică, a fost descris pentru prima dată de Hanes şi se caracterizează
prin: caracter autozomal recesiv, teleangiectazii multiple la nivelul
tegumentelor şi mucoaselor, manifestări gastrointestinale şi ale sistemului
nervos central.
Sindromul
afectează în special populaţia albă, dar poate fi întâlnit şi la pacienţi din
Asia sau Africa. Interesează în mod egal ambele sexe şi se manifestă în special
până în a treia decadă a vieţii.
Din
punct de vedere fiziopatologic, apariţia şi evoluţia sindromului ar putea fi
explicate prin mai multe ipoteze: degenerescenţa celulelor endoteliale; existenţa
unui deficit de joncţiune la nivelul celulelor endoteliale; existenţa unei
anomalii constituţionale a componentei elastice a peretelui arteriolar.
Diagnosticul
se pune pe prezenţa a mai mult de două din următoarele criterii: episoade
repetate de rinoragie; teleangiectazii prezente la nivelul tegumentelor şi mucoaselor; istorie familială; existenţa unor leziuni interne – teleangiectazii
gastrointestinale, malformaţii arteriovenoase.
mucoaselor; istorie familială; existenţa unor leziuni interne – teleangiectazii
gastrointestinale, malformaţii arteriovenoase.
Pacientul
poate prezenta numeroase teleangiectazii la nivelul tegumentelor feţei şi
corpului (fig. 4), mucoasei bucale (fig. 5) sau mucoasei foselor nazale.
Examenele
de laborator pot evidenţia o valoare scăzută a hemoglobinei, în urma sângerărilor
cronice, trombocitele pot avea valori normale sau pot fi uşor crescute, în timp
ce coagulograma este în limite normale.
Examenul
endoscopic nazal este cel care poate
depista existenţa teleangiectaziilor la nivelul mucoasei nazale, precum şi
sursa sângerării (fig. 6). Imagistica
(CT, IRM) poate evidenţia existenţa unor anomalii vasculo-nervoase.
Tratamentul poate fi medicamentos sau
chirurgical. Terapia medicamentoasă poate realiza profilaxia infecţioasă prin
administrarea antibioticelor. Chirurgical, se poate efectua electrocoagularea
sau fotocoagularea teleangiectaziilor de la nivelul mucoasei foselor nazale.
De asemenea, se poate efectua şi distrugerea leziunilor cu ajutorul laserului
CO2, Nd-YAG sau Argon. În cazul recidivelor, se poate ajunge la embolizarea
ramurilor arterei carotide externe sau la scleroza teleangiectaziilor. În
cazul sângerărilor rebele, se poate practica obturarea foselor nazale prin
sutura orificiilor narinare.
Sindromul
Sturge–Weber
sau angiomatoza encefalo-trigeminală reprezintă o boală congenitală rară,
cu dublă componentă – neurologică şi tegumentară. În literatura de specialitate
este enunţată o incidenţă a sindromului de 1 la 50.000 de pacienţi, ambele sexe
fiind afectate în mod egal.
Sindromul
apare în urma persistenţei unui plex vascular embrionar. Persistenţa plexului
duce la apariţia unui angiom, ce cuprinde leptomeningele, faţa şi ochiul
ipsilateral. Secundar, apare ischemie cerebrală, pierdere neuronală şi calcificări
la nivelul cortexului, aceste manifestări accentuând crizele convulsive.
Clinic,
sindromul se poate manifesta prin: pată roşie violacee apărută încă de la naştere
pe aria de inervaţie a nervului V; malformaţii vasculare meningeale
ipsilaterale; se poate asocia cu retard mintal, crize epileptice sau
convulsive, glaucom.
Terapia
medicamentoasă se adresează crizelor convulsive şi epileptice, prin administrarea
de medicamente anticonvulsivante. Se poate asocia terapia simptomatică pentru
glaucom sau cefalee. Tratamentul chirurgical este de ales în cazul crizelor
convulsive sau epileptice refractare, glaucomului.
Sindromul Maffucci, cunosut şi sub denumirea de hemangiom
cavernos multiplu, se caracterizează prin discondroplazie, cu deformarea şi
scurtarea oaselor afectate, leziuni vasculare viscerale în unele cazuri, şi se
poate asocia un condrosarcom (25% din cazuri).
Sindromul
Maffucci este o afecţiune rară, 160 de cazuri fiind raportate în literatura
engleză şi aproximativ 100 în SUA, leziunile fiind descrise cel mai frecvent
până la vârsta de 4–5 ani.
Pacienţii
sunt mici de înălţime, prezentând inegalitatea membrelor superioare sau/şi
inferioare din cauza anomaliilor osoase. Prezintă excrescenţe moi, albăstrui la
nivelul extremităţilor membrelor.
Monitorizarea
evoluţiei bolii se poate face prin imagistică, condrosarcomul fiind cea mai
frecventă formă de evoluţie a modificărilor osoase. Se urmăresc  elementele de
malignitate, cum ar fi: distrugerea corticală osoasă, zone de demineralizare
osoasă.
elementele de
malignitate, cum ar fi: distrugerea corticală osoasă, zone de demineralizare
osoasă.
În
cazul pacienţilor asimptomatici, nu se instituie tratament, aceştia fiind
monitorizaţi periodic. În momentul în care se suspicionează o transformare
malignă, se practică biopsia osoasă.
Boala
Von Hippel–Lindau se caracterizează prin asocierea de hemangioame
retiniene şi cerebeloase, chisturi renale şi chisturi pancreatice.
Iniţial,
majoritatea pacienţilor sunt asimptomatici, însă, pe măsură ce hemangioamele
retiniene cresc în dimensiuni, pacientul poate prezenta pierderea acuităţii
vizuale, dezlipire de retină, edem macular sau glaucom.
În ceea
ce priveşte hemangioamele cerebeloase, acestea se diagnostichează în jurul
vârstei de 25 de ani şi se pot manifesta prin diverse semne neurologice.
Prezenţa
chisturilor renale nu reprezintă o problemă majoră de sănătate pentru pacienţi,
dar capacitatea ridicată a acestora de malignizare este principala cauză de
mortalitate.
Monitorizarea
periodică reprezintă principala strategie terapeutică în cazul pacienţilor cu
boală Von Hippel–Lindau. În cazul evoluţiei chisturilor renale, se poate ajunge
la dializă sau la intervenţie chirurgicală, care poate consta în nefrectomie
uni- sau bilaterală.