Sindromul Ehlers-Danlos (SED) este un grup
eterogen de boli genetice rare, care afectează 1:2.500–1:5.000 de indivizi.
Este cauzat de un defect în structura, producerea sau prelucrarea colagenului
sau a proteinelor care interacţionează cu colagenul, cum ar fi mutaţii în
genele COL5A sau COL3A. Ca urmare, apar modificări în ţesutul conjunctiv;
fragilitatea pielii şi instabilitatea articulară sunt rezultatul cantităţii
reduse ori calităţii defectuoase a colagenului.
Caracteristicile clinice ale SED au fost
descrise pentru prima dată de Hipocrate (400 î.Hr.). Sindromul este numit după
medicii Edvard Ehlers (Danemarca) şi Henri-Alexandre Danlos (Franţa), care au
descris afecţiunea la începutul secolului 20.
Tablou clinic
Cele
mai frecvente semne ale SED sunt: hipermobilitate articulară,
hiperextensibilitate a pielii şi fragilitate a ţesuturilor. Există şase tipuri principale
de SED, clasificate în funcţie de manifestările clinice, semne şi simptome.
Fiecare tip este definit ca o tulburare distinctă, care se păstrează în cadrul
aceleiaşi familii.
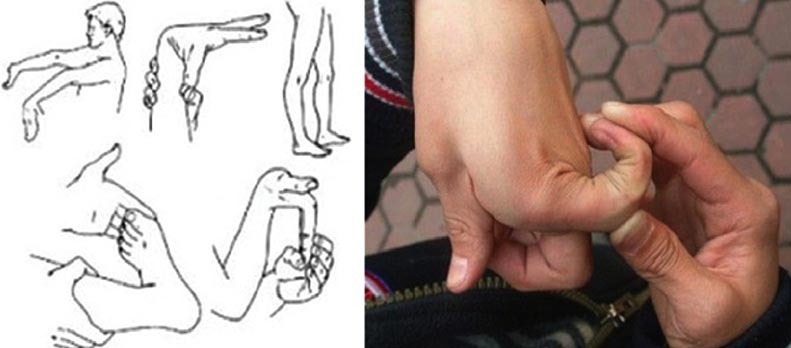
Hiperflexibilitate
articulară. Deoarece ţesutul
conjunctiv este mai relaxat, articulaţiile se pot deplasa mult dincolo de
limitele normale de mişcare (fig. 1).
Sunt afectate în special articulaţiile mici.
De asemenea, la nivelul articulaţiilor poate
apărea instabilitate, predispunând pacienţii la dislocări şi/sau subluxaţii
frecvente, dureri articulare sau debut precoce al osteoartritei.
Elasticitate
excesivă a pielii. Ţesutul
conjunctiv lax permite pielii să se întindă mai mult decât în mod normal (fig. 2). Aceasta este, de asemenea,
foarte catifelată.
Piele fragilă.Rănile
pielii se vindecă defectuos, cu cicatrici cheloide sau defecte de închidere.
Acumulări de
grăsime (puncte) în zonele de presiune.Pot apărea în jurul genunchilor sau coatelor, putând fi vizibile pe
radiografii.
Alte
semne mai puţin comune
asociate SED sunt: fragilitate crescută (rupturi) la nivel arterial, intestinal
sau uterin; scolioză la naştere şi fragilitate sclerală; tonus muscular slab;
prolaps de valvă mitrală; afecţiuni gingivale.
Severitatea simptomatologiei şi afectării
clinice variază de la o persoană la alta. Unii pacienţi pot avea
hiperflexibilitate articulară, dar fără simptome ale pielii.
Tipuri de sindrom Ehlers-Danlos
În
trecut, au existat 10 tipuri recunoscute de SED. În 1997, cercetătorii au
propus o clasificare mai simplă, care a redus numărul de tipuri majore de boală
la şase şi le-a dat nume descriptive. Pot exista şi alte tipuri de SED, dar au
fost raportate izolat în cadrul unor familii sau nu  au fost bine caracterizate.
Cu excepţia tipului 1, au fost identificate mutaţii specifice responsabile de
apariţia bolii. Rezultatele negative ale testelor genetice nu exclud
diagnosticul, deoarece nu au fost descoperite toate mutaţiile asociate fiecărui
tip de boală. Prin urmare, tabloul clinic este foarte important. Sunt rare
cazurile în care pacientul prezintă doar simptomatologia specifică unui anume
tip. Astfel, el poate asocia simptome din mai multe tipuri de boală, deşi
testele genetice pot fi pozitive pentru unul singur.
au fost bine caracterizate.
Cu excepţia tipului 1, au fost identificate mutaţii specifice responsabile de
apariţia bolii. Rezultatele negative ale testelor genetice nu exclud
diagnosticul, deoarece nu au fost descoperite toate mutaţiile asociate fiecărui
tip de boală. Prin urmare, tabloul clinic este foarte important. Sunt rare
cazurile în care pacientul prezintă doar simptomatologia specifică unui anume
tip. Astfel, el poate asocia simptome din mai multe tipuri de boală, deşi
testele genetice pot fi pozitive pentru unul singur.
În
ordinea prevalenţei în rândul populaţiei, tipurile de SED sunt:
1. Tipul
hipermobilitate (SED tipul III). Afectează 1:10.000 până la 1:15.000 de
indivizi şi se produce prin mecanism autozomal dominant sau recesiv – mutaţii
în oricare dintre genele COL3A1 şi TNXB. Hipermobilitatea este semnul distinctiv
al acestui tip, asociind manifestări mai puţin grave ale pielii. Sunt
caracteristice instabilitatea articulară şi durerile cronice
musculo-scheletice. Pacienţii prezintă frecvent dislocări şi subluxaţii
articulare, cu sau fără traume. Ca rezultat, durerea este un simptom comun; ea
este severă şi continuă. Osteoartrita este frecventă şi are debut precoce.
2. Tipul
clasic (SED tipurile I şi II). Afectează 1:20.000 până la 1:50.000 de
indivizi, prin mecanism autozomal dominant şi afectează colagenul tip V. Tipul
I de boală se asociază de obicei cu afectarea severă a pielii; tipul II – cu
afectare uşoară/moderată a acesteia. Pacienţii cu tipul clasic pot avea aceleaşi
simptome ca şi cei cu tipul III (hipermobilitate), principala diferenţă fiind
afectarea predominantă a pielii. Genele responsabile de acest tip de boală sunt
COL5A1, COL5A2, COL1A1.
3. Tipul
vascular (SED tipul IV). Este cauzat de un defect autozomal dominant
în sinteza de colagen tip III; afectează 1:100.000–1:250.000 de persoane. Este
considerat unul dintre cele mai grave forme de SED, deoarece vasele sanguine şi
organele sunt fragile şi predispuse la rupere. Mulţi pacienţi au un aspect
facial caracteristic (ochi mari, bărbie mică, obraji scofâlciţi, nas şi buze
subţiri, urechi fără lobi), o statură mică, constituţie subţire, piele subţire,
palidă, translucidă (vene vizibile de obicei la nivelul pieptului şi
abdomenului), cu hematoame şi echimoze (fără traume) (fig. 4). Aproximativ una din patru persoane cu SED tip vascular
are o problemă semnificativă de sănătate la 20 de ani şi mai mult de 80% vor
dezvolta complicaţii ameninţătoare de viaţă înainte de 40 de ani. Principala
genă responsabilă de apariţia acestui tip este COL3A1.
4. Tipul
cifoscolioză (SED tipul VI). Este cauzat de un defect recesiv autozomal,
deficitul enzimei lizil-hidroxilază; este foarte rar, cu mai puţin de 60 de
cazuri raportate în literatura de specialitate. Este caracterizat prin scolioză
progresivă, sensibilitate oculară şi slăbiciune musculară severă. Gena specifică
este PLOD1.
5. Tipul
artrochalazie (SED tipurile VII A şi B). Sunt aproximativ 30 de cazuri
raportate până în prezent. Afectează sinteza de colagen tip I. Se caracterizează
prin articulaţii foarte largi şi dislocări ale ambelor şolduri. Articulaţiile
sunt mult mai laxe, cu un grad de instabilitate de două ori mai mare decât
tipul III. Genele afectate sunt COL1A1 şi COL1A2.
6. Tipul
dermatosparaxis (SED tipul VII C). Este cel mai rar; se caracterizează
prin piele extrem de fragilă şi căzută. Gena afectată este ADAMTS2.
Diagnostic
Diagnosticul
se bazează în primul rând pe istoricul familial şi evaluarea clinică. Pacientul
este încadrat în funcţie de caracteristicile fiecărui tip de SED. Testarea
genetică este disponibilă pentru cinci tipuri de SED, cu excepţia celui mai
frecvent, cu hipermobilitate. Testele variază în precizie; în cele mai multe
cazuri, testarea genetică ar trebui să fie utilizată conservator pentru a
confirma diagnosticul, mai degrabă decât pentru a-l exclude.
 Teste
diagnostice: biopsia cutanată cu
evaluare la microscopul electronic – în tipul clasic evidenţiază o deformare în
conopidă a fibrelor de colagen (dar nu este o modificare specifică SED); teste
biochimice pe celule dermice (fibroblaste); teste genetice: secvenţiere genică,
analiza deleţiilor/duplicaţiilor, alelelor.
Teste
diagnostice: biopsia cutanată cu
evaluare la microscopul electronic – în tipul clasic evidenţiază o deformare în
conopidă a fibrelor de colagen (dar nu este o modificare specifică SED); teste
biochimice pe celule dermice (fibroblaste); teste genetice: secvenţiere genică,
analiza deleţiilor/duplicaţiilor, alelelor.
Diagnostic diferenţial
Diagnosticul
diferenţial se face cu alte afecţiuni cu simptome şi semne asemănătoare. Cutis laxa, sindromul Marfan, boala
Menkes, fibromialgia sunt cel mai frecvent menţionate. Anumite trăsături
clinice ale SED îl diferenţiază de alte afecţiuni similare: statura mică, ochi
mari, gura şi/sau bărbia mică; palatul poate avea un arc mare, provocând
aglomerări dentare, vasele sanguine pot fi, uneori, uşor vizibile prin pielea
translucidă, mai ales pe toracele anterior.
Tratament
Nu
există tratament specific, dar terapiile adjuvante contribuie la controlul
bolii şi prevenirea complicaţiilor.
Se
recomandă tratamentul durerii, al tensiunii arteriale (vasele fiind fragile,
reducerea stresului la nivelul acestora se realizează prin menţinerea unei
presiuni scăzute). Pentru prevenirea afectării articulare, este recomandată
fizioterapia cu exerciţii de consolidare musculară. În cazuri rare, intervenţiile
chirurgicale rezolvă pe cât posibil problemele cauzate de dislocaţii
articulare. Un rol foarte important îl are şi autoîngrijirea. Pacienţii sunt sfătuiţi
să evite traumatismele, sporturile de contact, să păstreze ordinea la domiciliu
pentru a preveni căderile şi leziunile, să utilizeze săpunuri pentru piele
sensibilă şi creme de protecţie solară, pentru a evita orice fel de agresiuni
ale pielii.
Sfat genetic
Din
cauza eterogenităţii genetice a bolii şi a mutaţiilor de novo, adesea sfatul genetic este dificil de acordat. În cazurile
clasice, cu transmitere conform unui model evident, riscul de recurenţă este
stabilit în funcţie de tipul transmiterii monogenice. Cel mai frecvent, acest
risc va fi de 1:2 în cazul transmiterii autozomal dominante şi 1:4 în formele
autozomal recesive.
Diagnostic prenatal
 Diagnosticul
molecular este posibil teoretic, dacă mutaţia a fost evidenţiată la părintele
afectat. În practică, acest test este încă aproape excepţional.
Diagnosticul
molecular este posibil teoretic, dacă mutaţia a fost evidenţiată la părintele
afectat. În practică, acest test este încă aproape excepţional.
Evoluţie şi prognostic Evoluţia
şi prognosticul diferă în funcţie de tipul şi gravitatea bolii. Există forme uşoare,
medii sau severe, care, în cursul evoluţiei, pot prezenta complicaţii grave. În
SED tip IV pot apărea perforaţii de colon, rupturi arteriale spontane, prolaps
de valvă mitrală. Sarcina sau naşterea (adesea prematură) se pot complica cu
ruptură uterină. În SED tip VI există risc de leziuni de cornee, subluxaţii de
cristalin sau decolare de retină.
Grupuri de sprijin
În
cadrul Centrului de Boli Rare NoRo din Zalău se oferă sprijin psiho-medical
pacienţilor şi familiilor acestora, prin întâlniri de grup. Pacienţii din toată
ţara sunt monitorizaţi şi instruiţi pentru a-şi controla cât mai corect boala.
La nivel internaţional, Fundaţia Ehlers-Danlos are o platformă web (http://www.ednf.org/) ce oferă sprijin şi
informare pentru pacienţii cu SED şi familiile lor.