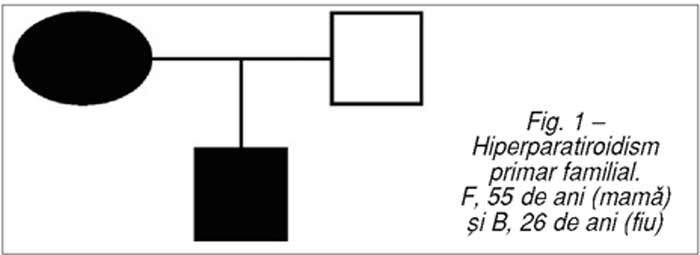
Hiperparatirodismul primar, cu o prevalenţă de 21 de cazuri la 100.000 de locuitori, este cea mai frecventă cauză de hipercalcemie. Pacientul se prezintă, deseori, la medic cu complicaţii renale (nefrocalcinoză, retenţie azotată, nefrolitiază etc.), cardiace (HTA, bradicardie sau QT scurt) sau neurologice (depresie). Prof. dr. Constantin Dumitrache şi dr. Irina Popescu prezintă, într-un amplu material, mecanismele şi dificultăţile clinice pe care le implică diagnosticul de hipercalcemie familială.
Sindromul
MEN2A este transmis autozomal dominant cu penetranţă genetică 95%.
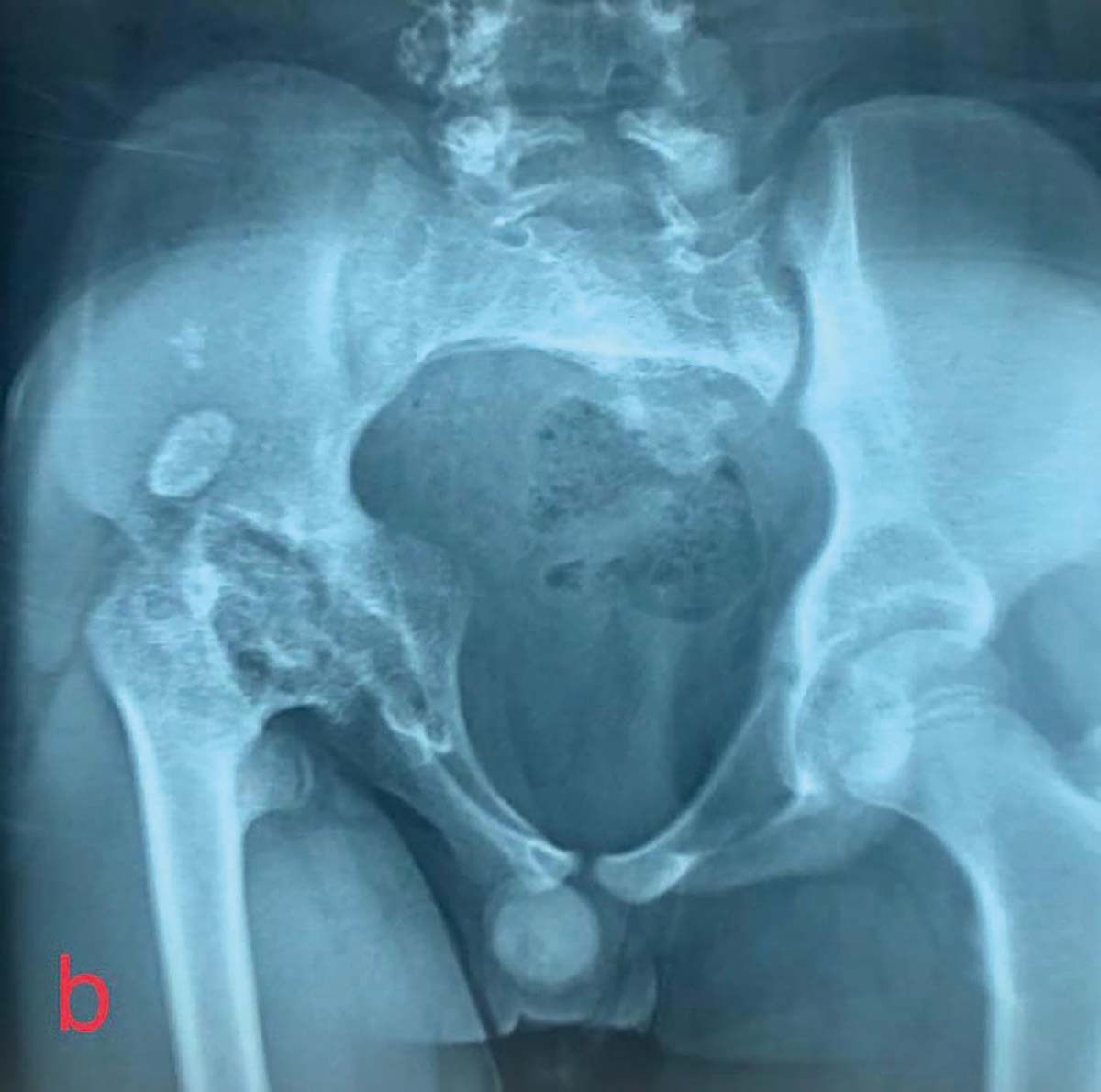
Hiperparatiroidismul primar prin hiperplazie paratiroidiană poliglandulară
apare doar la 25% din cazuri (5), carcinomul medular tiroidian şi
feocromocitomul fiind manifestări frecvente ale sindromului. Poate asocia
amiloidoză cutanată licheniformă şi boala Hirschprung. Este determinat de mutaţia
germinală activatoare a protooncogenei RET localizate la nivelul cromozomului
10q11.2 (9). Oncogena RET codifică un receptor tirozin-kinazic a cărui
stimulare promovează proliferarea celulară. Când este afectat codonul 634 al
genei RET, incidenţa hiperparatiroidismului este maximă (10).
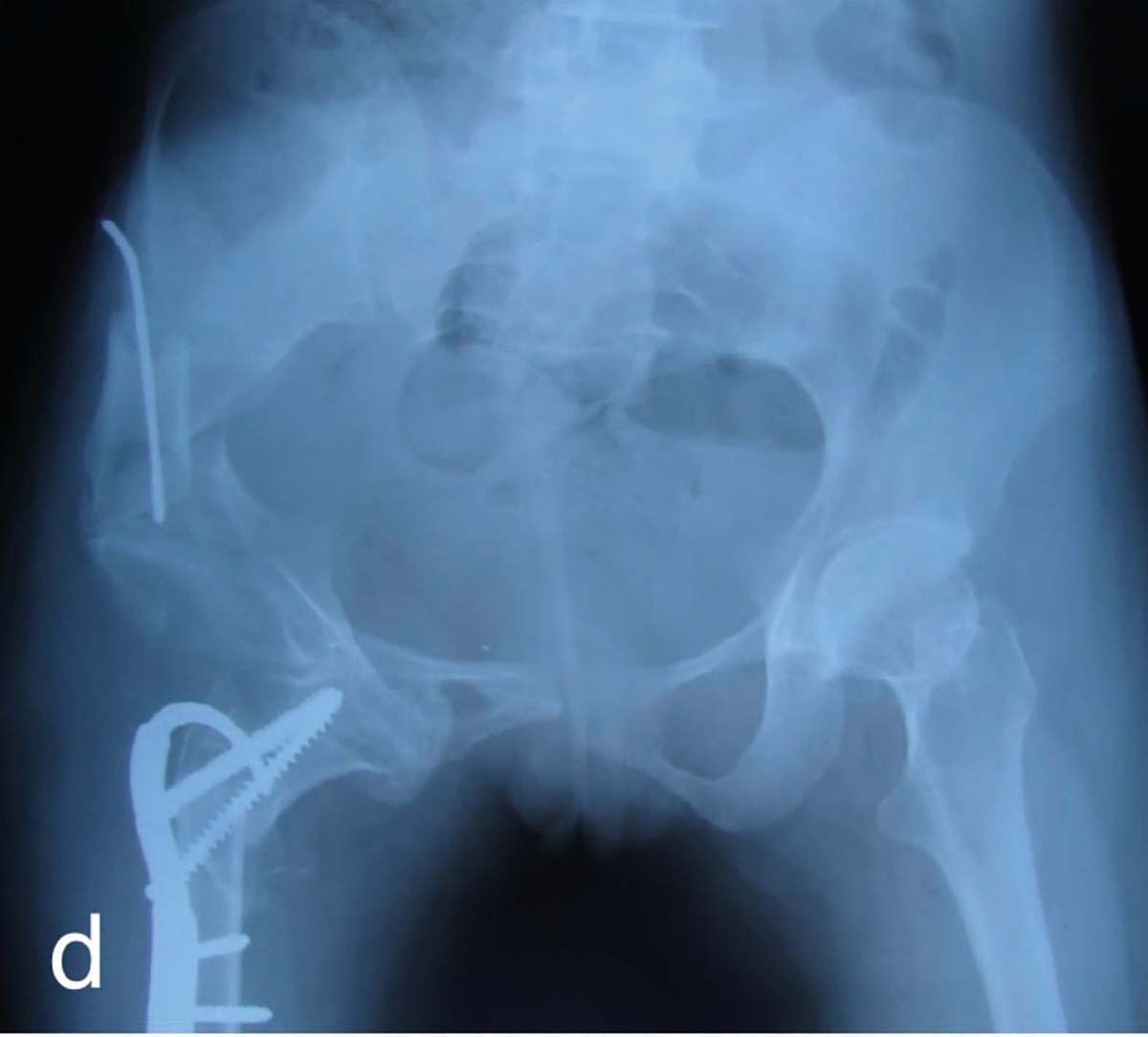
 izolat care dezvoltă carcinom paratiroidian.
izolat care dezvoltă carcinom paratiroidian. normetanefrinelor plasmatice şi urinare a fost normală.
Cortizolul plasmatic şi metaboliţii săi urinari (bază şi inhibiţie) au fost
normali, ca şi renina şi aldosteronul.
normetanefrinelor plasmatice şi urinare a fost normală.
Cortizolul plasmatic şi metaboliţii săi urinari (bază şi inhibiţie) au fost
normali, ca şi renina şi aldosteronul.Concluzii
Hipercalcemia familială constituie un grup heterogen de afecţiuni. Asemenea celorlalte forme de hipercalcemie, este adesea asimptomatică şi dozarea calciului în cadrul testelor de rutină este de importanţă vitală pentru diagnosticul precoce. Investigaţiile paraclinice complexe confirmă diagnosticul, iar tratamentul curativ este cel chirurgical. Testarea genetică ocupă un loc important în evaluarea pacientului, mai ales în situaţiile în care este necesară aprecierea riscului de apariţie a carcinomului paratiroidian, precum şi investigarea rudelor de sânge, impunând monitorizarea atentă a celor cu mutaţii confirmate.
Bibliografie
1. Wermers RA, Khosla S, Atkinson EJ, Achenbach SJ, Oberg AL, Grant CS, Melton LJ 3rd. Incidence of primary hyperparathyroidism in Rochester, Minnesota, 1993-2001: an update on the changing epidemiology of the disease. J Bone Miner Res. 2006 Jan;21(1):171-7
2. Fraker DL, Harsono H, Lewis R. Minimally invasive parathyroidectomy: benefits and requirements of localization, diagnosis, and intraoperative PTH monitoring. long-term results. World J Surg. 2009 Nov;33(11):2256-65
3. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA Jr, Marx SJ. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001 Dec;86(12):5658-71
4. Bilezikian JP, Brandi ML, Rubin M, Silverberg SJ. Primary hyperparathyroidism: new concepts in clinical, densitometric and biochemical features. J Intern Med. 2005 Jan;257(1):6-17
5. Gardner DG, Shoback D. Greenspan’s Basic and Clinical Endocrinology. Chapter 23, McGraw Hill Medical 2007
6. Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjöld M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988 Mar 3;332(6159):85-7
7. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997 Apr 18;276(5311):404-7
8. Szabó J, Heath B, Hill VM, Jackson CE, Zarbo RJ, Mallette LE, Chew SL, Besser GM, Thakker RV, Huff V, et al. Hereditary hyperparathyroidism-jaw tumor syndrome: the endocrine tumor gene HRPT2 maps to chromosome 1q21-q31. Am J Hum Genet. 1995 Apr;56(4):944-50
9. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Höfler H, Fend F, Graw J, Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006 Oct 17;103(42):15558-63
10. Simonds WF, James-Newton LA, Agarwal SK, Yang B, Skarulis MC, Hendy GN, Marx SJ. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore). 2002 Jan;81(1):1-26
11. Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999 Jan 8;96(1):143-52
12. Larsson C. Dissecting the genetics of hyperparathyroidism—new clues from an old friend. J Clin Endocrinol Metab. 2000 May;85(5):1752-4
13. Simonds WF, James-Newton LA, Agarwal SK, Yang B, Skarulis MC, Hendy GN, Marx SJ. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore). 2002 Jan;81(1):1-26
14. Marx SJ, Simonds WF, Agarwal SK, Burns AL, Weinstein LS, Cochran C, Skarulis MC, Spiegel AM, Libutti SK, Alexander HR Jr, Chen CC, Chang R, Chandrasekharappa SC, Collins FS. Hyperparathyroidism in hereditary syndromes: special expressions and special managements. J Bone Miner Res. 2002 Nov;17 Suppl 2:N37-43
15. Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, Agarwal SK, Sood R, Jones MP, Moses TY, Haven C, Petillo D, Leotlela PD, Harding B, Cameron D, Pannett AA, Höög A, Heath H 3rd, James-Newton LA, Robinson B, Zarbo RJ, Cavaco BM, Wassif W, Perrier ND, Rosen IB, Kristoffersson U, Turnpenny PD, Farnebo LO, Besser GM, Jackson CE, Morreau H, Trent JM, Thakker RV, Marx SJ, Teh BT, Larsson C, Hobbs MR. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002 Dec;32(4):676-80
16. Shattuck TM, Välimäki S, Obara T, Gaz RD, Clark OH, Shoback D, Wierman ME, Tojo K, Robbins CM, Carpten JD, Farnebo LO, Larsson C, Arnold A. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003 Oct 30;349(18):1722-9
17. Simonds WF, Robbins CM, Agarwal SK, Hendy GN, Carpten JD, Marx SJ. Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab. 2004 Jan;89(1):96-102
18. Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993 Dec 31;75(7):1297-303
19. Fuleihan GE, Brown EM, Heath HH III. Familial benign hypocalciuric hypercalcemia and neonatal primary hyperparathyroidism. In: Principles of Bone Biology, 3rd Edition (Bilezikian JP, Raisz LG, Martin TJ, eds). Academic Press, San Diego, 2008:1327-45
20. Arnold A. Familial hyperparathyroidism. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism (Rosen CJ, ed), 7th edition. American Society for Bone and Mineral Research, Washington, DC, 2008:361-7
21. Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, Bilezikian JP. Parathyroid carcinoma. J Bone Miner Res. 2008 Dec;23(12):1869-80
22. Teh BT, Farnebo F, Twigg S, Höög A, Kytölä S, Korpi-Hyövälti E, Wong FK, Nordenström J, Grimelius L, Sandelin K, Robinson B, Farnebo LO, Larsson C. Familial isolated hyperparathyroidism maps to the hyperparathyroidism-jaw tumor locus in 1q21-q32 in a subset of families. J Clin Endocrinol Metab. 1998 Jun;83(6):2114-20
23. Kassem M, Kruse TA, Wong FK, Larsson C, Teh BT. Familial isolated hyperparathyroidism as a variant of multiple endocrine neoplasia type 1 in a large Danish pedigree. J Clin Endocrinol Metab. 2000 Jan;85(1):165-7
24. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, Lewis-Barned N, McCredie D, Powell H, Kendall-Taylor P, Brown EM, Thakker RV. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996 Oct 10;335(15):1115-22
25. Carling T, Szabo E, Bai M, Ridefelt P, Westin G, Gustavsson P, Trivedi S, Hellman P, Brown EM, Dahl N, Rastad J. Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J Clin Endocrinol Metab. 2000 May;85(5):2042-7
26. Cascón A, Huarte-Mendicoa CV, Javier Leandro-García L, Letón R, Suela J, Santana A, Costa MB, Comino-Méndez I, Landa I, Sánchez L, Rodríguez-Antona C, Cigudosa JC, Robledo M. Detection of the first gross CDC73 germline deletion in an HPT-JT syndrome family. Genes Chromosomes Cancer. 2011 Nov;50(11):922-9
27. Frank-Raue K, Leidig-Bruckner G, Haag C, Schulze E, Lorenz A, Schmitz-Winnenthal H, Raue F. Inactivating calcium-sensing receptor mutations in patients with primary hyperparathyroidism. Clin Endocrinol (Oxf). 2011 Mar 29
28. Motokura T, Bloom T, Kim HG, Jüppner H, Ruderman JV, Kronenberg HM, Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991 Apr 11;350(6318):512-5
29. Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC, Manickam P, Olufemi SE, Skarulis MC, Doppman JL, Alexander RH, Kim YS, Saggar SK, Lubensky IA, Zhuang Z, Liotta LA, Chandrasekharappa SC, Collins FS, Spiegel AM, Burns AL, Marx SJ. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet. 1997 Aug;16(4):375-8
30. Fukuda N, Tanaka H, Tominaga Y, Fukagawa M, Kurokawa K, Seino Y. Decreased 1,25-dihydroxyvitamin D3 receptor density is associated with a more severe form of parathyroid hyperplasia in chronic uremic patients. J Clin Invest. 1993 Sep;92(3):1436-43
31. Sudhaker Rao D, Han ZH, Phillips ER, Palnitkar S, Parfitt AM. Reduced vitamin D receptor expression in parathyroid adenomas: implications for pathogenesis. Clin Endocrinol (Oxf). 2000 Sep;53(3):373-81
32. Carling T, Kindmark A, Hellman P, Holmberg L, Akerström G, Rastad J. Vitamin D receptor alleles b, a, and T: risk factors for sporadic primary hyperparathyroidism (HPT) but not HPT of uremia or MEN 1. Biochem Biophys Res Commun. 1997 Feb 13;231(2):329-32
33. Carling T, Ridefelt P, Hellman P, Rastad J, Akerström G. Vitamin D receptor polymorphisms correlate to parathyroid cell function in primary hyperparathyroidism. J Clin Endocrinol Metab. 1997 Jun;82(6):1772-5
34. Carling T, Rastad J, Akerström G, Westin G. Vitamin D receptor (VDR) and parathyroid hormone messenger ribonucleic acid levels correspond to polymorphic VDR alleles in human parathyroid tumors. J Clin Endocrinol Metab. 1998 Jul;83(7):2255-9
35. Correa P, Rastad J, Schwarz P, Westin G, Kindmark A, Lundgren E, Akerström G, Carling T. The vitamin D receptor (VDR) start codon polymorphism in primary hyperparathyroidism and parathyroid VDR messenger ribonucleic acid levels. J Clin Endocrinol Metab. 1999 May;84(5):1690-4
Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Titularii abonamentelor pe 12 luni sunt creditați astfel de:

Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Află mai multe informații despre oferta de abonare.
Cookie-urile ne ajută să vă îmbunătățim experiența pe site-ul nostru. Prin continuarea navigării pe site-ul www.viata-medicala.ro, veți accepta implicit folosirea de cookie-uri pe parcursul vizitei dumneavoastră.
Da, sunt de acord Aflați mai multe