Boala
Huntington (BH) este o afecţiune neurologică ereditară rară, determinată
monogenic, care debutează tardiv şi se manifestă prin tulburări de coordonare a
mişcărilor, degradare cognitivă progresivă (demenţă) şi tulburări psihotice
cauzate de moartea selectivă a neuronilor. Denumită şi choree Huntington, a
fost descrisă pentru prima oară de George Huntington, în 1872. Afectează
aproximativ 1 din 20.000 de locuitori în Europa, dar este mai rară la alte
populaţii.
Aspecte
genetice.
Deşi este vorba despre o afecţiune ereditară monogenică, în general boala
debutează tardiv, între 35 şi 45 de ani (există însă forme cu debut în copilărie
sau numai după 60 de ani).
Moartea
apoptotică selectivă a neuronilor (sau după studii mai noi, întreruperea procesării
informaţiei între neuroni) este indusă de secvenţa poliglutaminică amplificată
datorită unei mutaţii dinamice a genei IT15 (4p16.3), care codifică
huntingtina.
Mutaţia
dinamică, un mecanism recunoscut şi elucidat abia în anii ’90, spre deosebire
de mutaţiile clasice prezintă o serie de trăsături neobişnuite. La începutul
genei există o secvenţă repetitivă (vezi
tabelul), conţinând la persoanele sănătoase 11–34 de repetări de CAG (cele
trei nucleotide: citozină, adenină, guanină codifică glutamina din proteina
huntingtină). Cu ocazia diviziunii celulare, numărul repetiţiilor acestor
nucleotide poate creşte (expansiune). Creşterea peste 35 a numărului de repetări
determină starea de boală, care este cu atât mai severă şi debutează cu atât
mai precoce cu cât numărul repetiţiilor este mai mare (fenomen de anticipaţie).
Această expansiune se poate produce şi cu ocazia meiozei la pacienţi, ceea ce
determină moştenirea de către copil a unei secvenţe mai lungi faţă de cea
existentă la părinte, şi astfel o manifestare mai gravă a bolii. Expansiunea
secvenţei repetitive se produce în general în meioza masculină, astfel penetranţa
este mai mare când boala se transmite pe linie paternă. Celulele afectate de
această mutaţie dinamică sunt cele din creier, în special neuronii din nucleii
bazali, a căror moarte determină simptomatologia caracteristică bolii.
glutamina din proteina
huntingtină). Cu ocazia diviziunii celulare, numărul repetiţiilor acestor
nucleotide poate creşte (expansiune). Creşterea peste 35 a numărului de repetări
determină starea de boală, care este cu atât mai severă şi debutează cu atât
mai precoce cu cât numărul repetiţiilor este mai mare (fenomen de anticipaţie).
Această expansiune se poate produce şi cu ocazia meiozei la pacienţi, ceea ce
determină moştenirea de către copil a unei secvenţe mai lungi faţă de cea
existentă la părinte, şi astfel o manifestare mai gravă a bolii. Expansiunea
secvenţei repetitive se produce în general în meioza masculină, astfel penetranţa
este mai mare când boala se transmite pe linie paternă. Celulele afectate de
această mutaţie dinamică sunt cele din creier, în special neuronii din nucleii
bazali, a căror moarte determină simptomatologia caracteristică bolii.
Semne
clinice.
Manifestările clinice debutează în general între 35 şi 50 de ani şi se caracterizează prin următoarele tulburări
neurologice, cognitive şi comportamentale:
• tulburări
motorii de coordonare a mişcărilor, mers instabil, mişcări tipice spasmodice
necontrolate (mişcări rapide, neaşteptate, bizare ale feţei, membrelor sau
corpului, grimase, choree – dans);
• pierderea
progresivă a capacităţilor mintale, a funcţiilor cognitive, tulburări de memorie,
pierderea capacităţii de judecată, demenţă;
• tulburări
de vorbire;
• tulburări
de înghiţire;
• modificări
comportamentale, schimbări de personalitate, iritabilitate, instabilitate emoţională,
manifestări  antisociale;
antisociale;
• manifestări
psihotice, depresie, halucinaţii, confuzie, agitaţie, psihoze;
• crize
epileptiforme, în special în cazul formelor cu debut în copilărie.
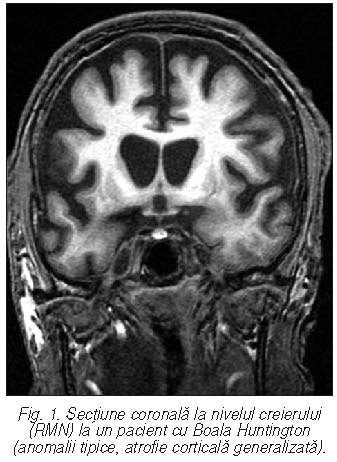
Diagnostic. În baza examenului
clinic neurologic şi a antecedentelor familiale se poate stabili diagnosticul.
Unele examinări paraclinice imagistice sunt
sugestive pentru boală (de ex. tomografia computerizată craniană poate arăta
atrofie cerebrală, fig. 1).
Microscopia electronică furnizează imagini ale neuronilor cu incluziuni
specifice bolii (fig. 2).
Analiza mutaţiei genice confirmă diagnosticul
clinic, aduce informaţii suplimentare în ceea ce priveşte prognosticul
(anticipaţie în funcţie de numărul repetiţiilor de trinucleotide) şi permite
diagnosticul la rudele sănătoase dar purtătoare de mutaţie care vor dezvolta
boala ulterior (diagnostic molecular presimptomatic).
Consult genetic. Boala Huntington se
transmite după modelul autozomal dominant, ceea ce înseamnă că dacă un părinte
este afectat, riscul de apariţie a bolii la copii este de 50%. Mutaţia poate apărea
şi de novo, situaţie în care părinţii
nu sunt afectaţi, dar boala se moşteneşte la descendenţii pacientului cu acelaşi
risc crescut, caracteristic transmiterii autozomal dominante.
Boala se transmite la descendenţii de sex
masculin sau feminin, în egală măsură, realizând în familie aşa-numita
transmitere pe verticală, cu mai multe generaţii succesive afectate (aspect
asemănător arborelui genealogic din fig.
3). Toate aceste aspecte complexe susţin necesitatea unui consult genetic
individualizat.
Diagnostic prenatal. Deşi este posibil,
oportunitatea diagnosticului presimptomatic şi prenatal constituie încă o dilemă
etică.
Evoluţie şi prognostic. După debutul clinic,
boala Huntington prezintă evoluţie progresivă. Însă rapiditatea evoluţiei şi
gravitatea simptomelor pot prezenta variaţii de la un caz la altul, chiar între
membrii aceleaşi familii.
În general, decesul se produce după 15–20 ani
de la debut, frecvent prin infecţii sau suicid.
Posibilităţi de tratament, îngrijire şi urmărire.În
prezent, BH se consideră incurabilă.
Intervenţiile terapeutice au ca scop
încetinirea progresiei şi menţinerea pentru o perioadă cât mai lungă a capacităţilor
motorii şi intelectuale.
Alegerea medicaţiei depinde de simptomele
prezente la pacienţi. Astfel, de exemplu, unele medicamente cu efect
antidopaminergic (de ex. haloperidol, fenotiazină) pot reduce tuburările
comportamentale şi motorii, iar altele se pot folosi pentru controlul mişcărilor
coreiforme (tetrabenazină, amantadină). Se pare că administarea coenzimei Q10
poate încetini progresia bolii.
Acordarea sfatului genetic constituie parte
integrantă a îngrijirii familiilor afectate de BH. Posibilitatea diagnosticului
presimptomatic poate fi oferit rudelor de vârstă adultă. Având în vedere riscul
mare de transmitere a bolii de 50%, pot fi luate în considerare alternative
reproductive (de exemplu, adopţia).
adultă. Având în vedere riscul
mare de transmitere a bolii de 50%, pot fi luate în considerare alternative
reproductive (de exemplu, adopţia).
Viaţa cotidiană. De la debutul bolii
pot fi necesare diferite măsuri de monitorizare şi supraveghere a pacienţilor,
care au obiective variate specifice cazurilor, de la utilizarea unor ajutoare
pentru memorizare până la prevenirea tentativelor de suicid.
Progresiv, pacienţii îşi pierd capacitatea de
autoîngrijire şi de comunicare şi necesită îngrijire, frecvent 24 de ore/zi în
stadiile avansate.
Alăturarea la un grup de suport alcătuit din
alţi pacienţii şi rude din familii afectate poate reduce stresul şi temerile
legate de boală şi ajuta în luarea unor decizii.